最近有粉丝提问,他的差异分析跟一篇nature文章的差异分析结果交集少得可怜,不知道自己哪里错误了。
数据集为TCGA-DLBC, 共48个样本,这个nature文献是根据AMBRA1和CCND3基因是否发生突变/扩增/deletion,将所有样本分为实验组和对照组(两个基因均没有发生任何variation的设为对照组);所以分组情况是 42 control+6 突变组
然后粉丝是 使用的DEGs方法为edgeR包,输入count数据,基因排除标准为sum==0,阈值为logFC 绝对值大于1 , p值<0.05, 最后取上下调共top300个基因。跟作者的top300取交集,只有34个。
一般来说这样的转录组差异分析提问,我会找对方要三张图。
转录组差异分析需要质量控制3张图
我在生信技能树的教程:《你确定你的差异基因找对了吗?》提到过,必须要对你的转录水平的全局表达矩阵做好质量控制,最好是看到标准3张图:
- 左边的热图,说明我们实验的两个分组,normal和npc的很多基因表达量是有明显差异的
- 中间的PCA图,说明我们的normal和npc两个分组非常明显的差异
- 右边的层次聚类也是如此,说明我们的normal和npc两个分组非常明显的差异
如果分组在3张图里面体现不出来,实际上后续差异分析是有风险的。这个时候需要根据你自己不合格的3张图,仔细探索哪些样本是离群点,自行查询中间过程可能的问题所在,或者检查是否有其它混杂因素,都是会影响我们的差异分析结果的生物学解释。
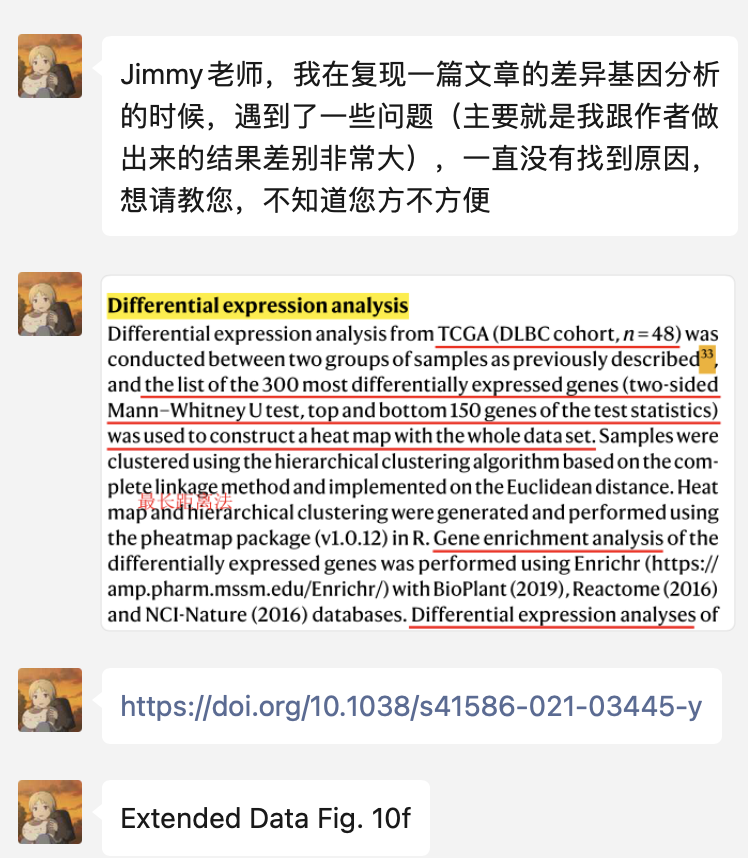
我看了看粉丝提到的 这个 数据集为TCGA-DLBC, 共48个转录组样本,分组情况是 42 野生型对照组 + 6 突变组 ,得到的3张图就很奇怪!
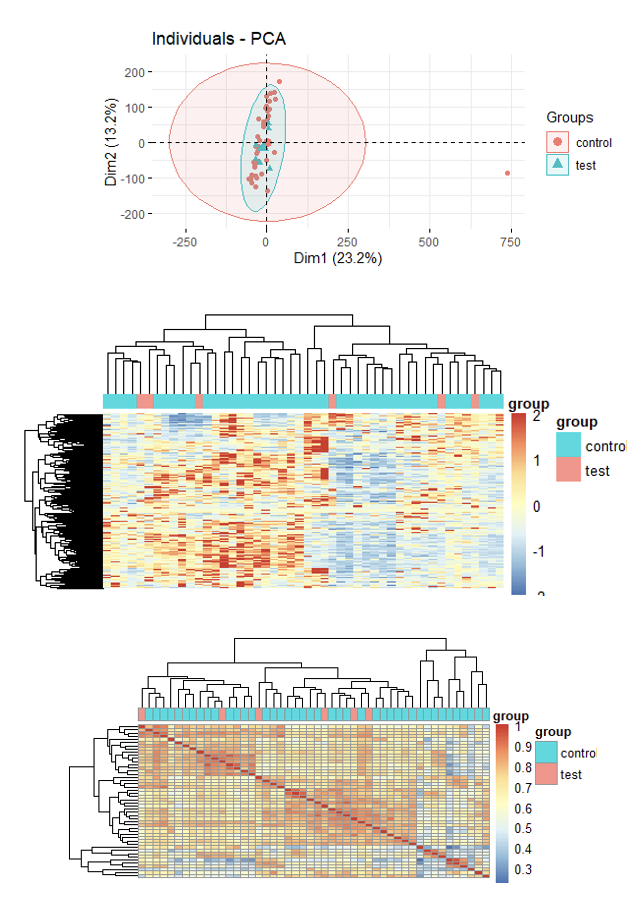
可以看到,这样的 42 野生型对照组 + 6 突变组 是完全随机的临床样品了,并不是标准的分组对照实验设计,没有所谓的组间差异大于组内差异的说法。两个分组的不同样品在各种质量控制图表里面都出现不同程度的混入,这样就是标准的强项找差异!如果你想看标准的三张图,比如发表于2021年9月27日,美国康奈尔医学院周乔课题组在Cell Stem Cell 期刊,文章标题是:《SATB2 preserves colon stem cell identity and mediates ileum-colon conversion via enhancer remodeling》,在线阅读链接 是:https://doi.org/10.1016/j.stem.2021.09.004 在附件就提到了这样的三张图:
因为本文里面的 42 野生型对照组 + 6 突变组 是强行分组, 所以差异分析的结果火山图热图也非常奇怪:
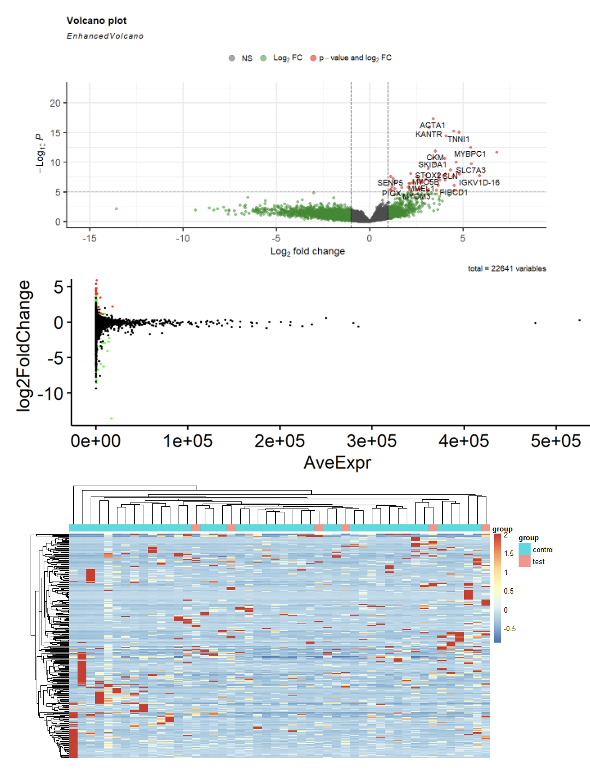
既然是强行找差异,使用标准差异分析流程处理它我们就不可能对漂亮的结果有什么期待了!
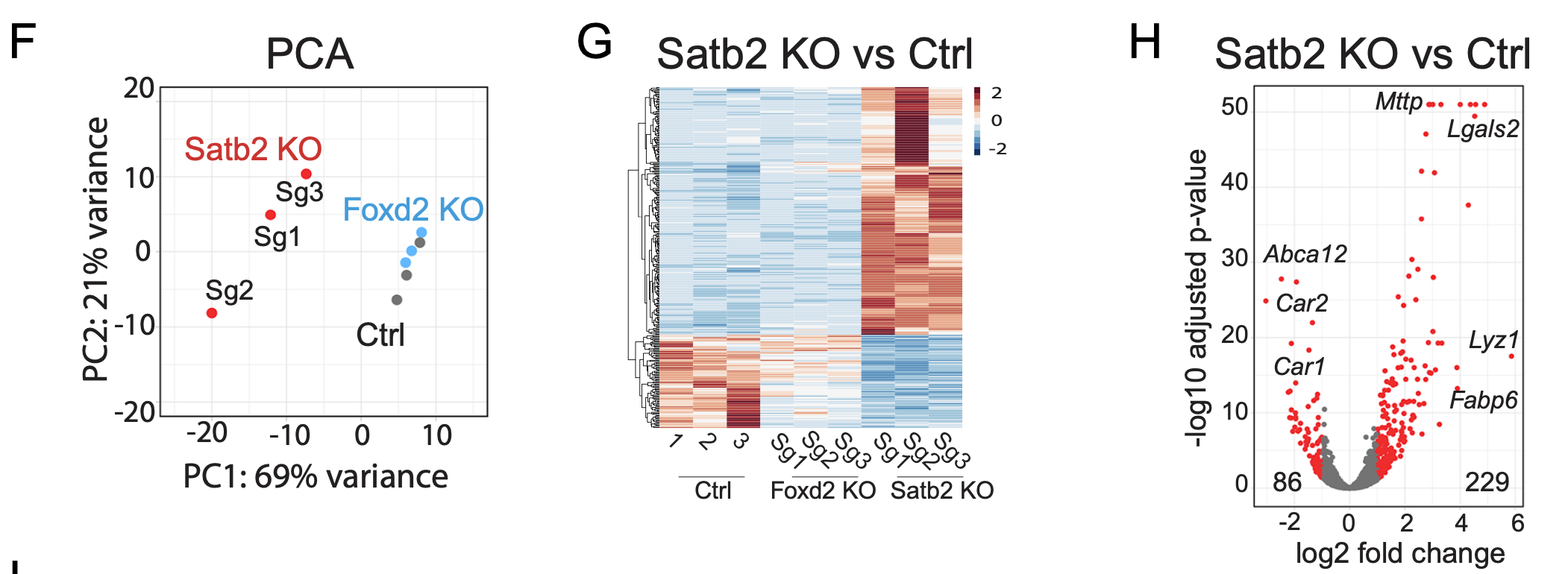
但是作者原文漂亮很多,他用Mann-Whitney U test找的差异基因,然后他的实验组聚类最后聚到一起了!如下所示:
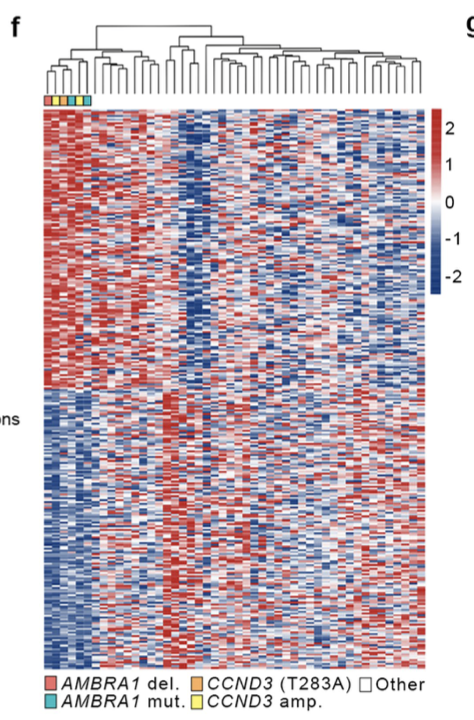
可以看到,其实作者找到的是一个分组,可以把48个病人分成比较好的两个分组,而且恰好作者关心的6个突变样品落入了其中的某一个分组, 这样的差异分析结果很有意思。
其实如果是强行分组,可以借鉴代谢组学里面的 的 PLS-DA 展示 方法,我们常规的PCA是无监督的降维方式,我们的表达量矩阵各自的样品是什么关系就展现什么关系,后续的分组仅仅是去给样品标记颜色而已,它是独立于这个无监督的PCA降维,所以如果你的样品并不是跟分组信息匹配,就容易出现混乱的PCA图,而不是泾渭分明的2个或者 多个分组。但是当无监督(PCA)无法很好地区分组间样本时,PLS-DA可以实现有效分离。并且PLS-DA和OPLS-DA所构建的分类预测模型,可进一步用于识别更多的样本类别,这是探索性的PCA方法无法做到的。 也就是说 PLS-DA 其实就是预先给定了分组信息,一定要找到分组的差异然后去进行展现,这样它们在图上就很容易看到已知的分组信息的泾渭分明啦。
虽然这样的nature正刊的差异分析是槽点满满,但 它毕竟是 补充图表内容,而且是 Extended Data Fig. 10f
不得不说,发一个nature正刊工作量真多
我们仅仅是讨论 Extended Data Fig. 10f 就这么多知识点了。他们首先需要使用公共数据库来说明他们研究的这两个基因的重要性:
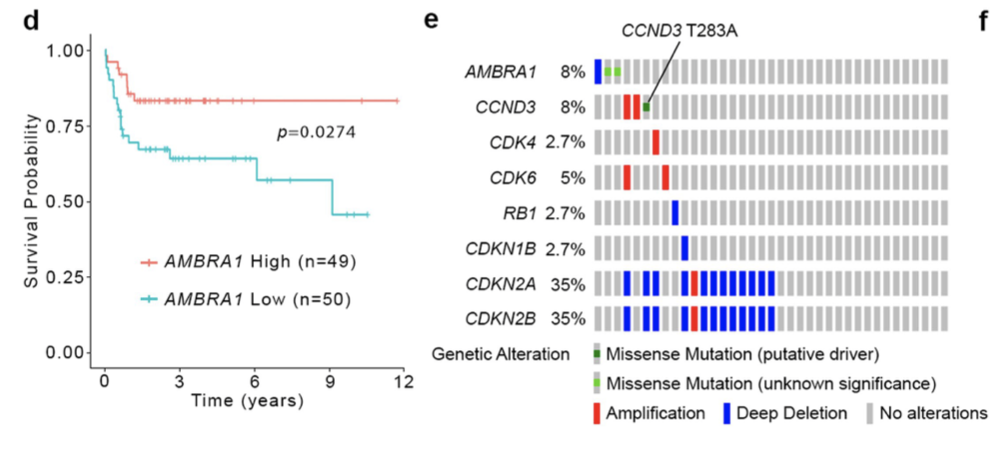
我在生信技能树多次分享过生存分析的细节;
- 人人都可以学会生存分析(学徒数据挖掘)
- 学徒数据挖掘之谁说生存分析一定要按照表达量中位值或者平均值分组呢?
- 基因表达量高低分组的cox和连续变量cox回归计算的HR值差异太大?
- 学徒作业-两个基因突变联合看生存效应
- TCGA数据库里面你的基因生存分析不显著那就TMA吧
- 对“不同数据来源的生存分析比较”的补充说明
- 批量cox生存分析结果也可以火山图可视化
- 既然可以看感兴趣基因的生存情况,当然就可以批量做完全部基因的生存分析
- 多测试几个数据集生存效应应该是可以找到统计学显著的!
- 我不相信kmplot这个网页工具的结果(生存分析免费做)
- 为什么不用TCGA数据库来看感兴趣基因的生存情况
- 200块的代码我的学徒免费送给你,GSVA和生存分析
- 集思广益-生存分析可以随心所欲根据表达量分组吗
- 生存分析时间点问题
- 寻找生存分析的最佳基因表达分组阈值
- apply家族函数和for循环还是有区别的(批量生存分析出图bug)
- TCGA数据库生存分析的网页工具哪家强
- KM生存曲线经logRNA检验后也可以计算HR值
生存分析是目前肿瘤等疾病研究领域的点睛之笔!
文章里面的突变与否分成两组,差异分析后,还需要对上下调基因进行生物学功能数据库富集:

文章的描述是 :g, Gene enrichment analysis of the top 300 most differentially expressed genes in patients harbouring alterations in AMBRA1 and CCND3 from TCGA (DLBC cohort) was performed using Enrichr (https://amp.pharm.mssm.edu/Enrichr/) with BioPlant (2019), Reactome (2016) and NCI-Nature (2016) databases.
如果大家对数据挖掘的中间过程的合理性不好把握,建议看完我两年前带学徒的时候,安排他们做的文献关键图表复现作业系列笔记分享,如下:
第一期(2018年秋季)
- 保姆式GEO数据挖掘演示—重现9分文章
- GEO数据挖掘-第一期-胶质母细胞瘤(GBM)
- GEO数据挖掘-第二期-三阴性乳腺癌(TNBC)
- GEO数据挖掘-第三期-口腔鳞状细胞癌(OSCC),WGCNA
- GEO数据挖掘-第四期-肝细胞癌(HCC),WGCNA
- GEO数据挖掘-第五期-肝细胞癌(HCC)-多组分开差异分析
- GEO数据挖掘-第六期-RNA-seq数据也照挖不误
- TCGA数据库的TP53突变型和TP53野生型BRCA病人的差异分析结果
- GEO数据库的耐药与敏感的患者组织内的成纤维细胞比较
- TCGA数据库中三阴性乳腺癌在亚洲人群中的差异表达
- TCGA数据库的有PIK3CA基因突变的肿瘤病人的转录水平变化
- TCGA数据库里面的乳腺癌的芯片表达数据进行差异分析
第二期(2019年全年)
- 1. 公共数据辅助乳腺癌的免疫治疗机制研究
- 2. 有生物学意义的复杂热图
- 3. 干扰MYC‑WWP1通路重新激活PTEN的抑癌活性——3步搞定GSEA分 析
- 4. 按基因在染色体上的顺序画差异甲基化热图
- 5. 热图、⻙恩图、GO富集分析图(有了转录组数据不知道该怎么写⽂ 章,看我就对了!)
- 6. 纯R代码实现ssGSEA算法评估肿瘤免疫浸润程度
- 7. 肿瘤异质性+免疫浸润细胞数据挖掘(可能是最简单的3分⽂章了)
- 8. ArrayExpress数据库的基因芯⽚原始数据处理,3D主成分图及聚类热 图
- 9. 学徒数据挖掘第⼆期汇总之多分组基因注释代码⼤放送
- 10. TCGA数据辅助甲基化区域的功能研究
- 11. 你确定你的差异基因找对了吗?
- 12. 看nature⽂章是如何设计和使⽤普通转录组数据
- 13. 不⼀定正确的多分组差异分析结果热图展现
- 14. 如果传统bulk转录组数据队列⾜够⼤也可以使⽤单细胞流程
- 15. 最简单的芯⽚挖掘也会出错(菜⻦团周⼀数据挖掘专栏第?期)
- 16. 乳腺癌的IHC分类和PAM50分型的差异情况
学徒作业
- 针对这个数据集,TCGA-DLBC, 共48个样本
- 首先去cbioportal拿到如上所示的指定基因突变结果全景图
- 然后根据AMBRA1和CCND3基因是否发生突变/扩增/deletion找到6个突变病人
- 然后针对 这个 数据集为TCGA-DLBC, 共48个转录组样本,分组情况是 42 野生型对照组 + 6 突变组 进行标准差异分析
- 然后针对 42 野生型对照组 + 6 突变组 进行 Mann-Whitney U test找的差异基因
- 绘制两个不同差异分析的热图