前面的教程 不同癌症的差异难道大于其与正常对照差异吗,以及 大样本量多分组表达量矩阵分析你难道没想到单细胞吗,我们认识了TCGA数据库的33种癌症的全部的表达量矩阵,并且格式化保存为了 Rdata 文件。并且批量走了estimate算法,得到了各个样品的基质细胞和免疫细胞的比例的打分值!
而且简单的可视化可以看到不同癌症内部的estimate的两个打分值是有异质性的,是连续值比较方便的做法是根据中位值在各个肿瘤分组内部划分高低组,然后批量进行差异分析!
首先载入全部的表达量矩阵和estimate的两个打分值结果
rm(list=ls())
options(stringsAsFactors = F)
library(stringr)
library(data.table)
library(Seurat)
load(file = 'sce.Rdata')
gp=substring(colnames(sce),14,15)
table(gp)
sce@meta.data$gp=gp
pd_mat=sce@assays$RNA@counts
pd_mat[1:4,1:4]
dat=log2(edgeR::cpm(pd_mat)+1)
# 首先载入针对seurat对象的 estimate 结果
pro='estimate_for_seurat'
load(file = file.path('estimate_results',
paste0('estimate_RNAseq-score-for-',
pro,'.Rdata')))
cg_scores=scores[as.numeric(scores$group )< 11,]
head(cg_scores)
因为是在各个肿瘤内部进行estimate的两个打分值结果高低分组,所以需要删除全部的正常对照组织的转录组测序表达量矩阵。
estimate的两个打分值结果高低分组并且走limma的voom算法的差异分析
针对转录组测序表达量矩阵我们通常是有3种差异分析算法,这里我们选择limma的voom算法,代码如下:
suppressMessages(library(limma))
suppressMessages(library(edgeR))
tp=unique(cg_scores$type)
stromal_DEG_limma_voom_list <- lapply(tp, function(i){
# i=tp[1]
tp_scores=cg_scores[cg_scores$type==i,]
group_list=ifelse(tp_scores$StromalScore > median(tp_scores$StromalScore ),'high','low')
exprSet=pd_mat[,match(gsub('[.]','-',rownames(tp_scores)),
colnames(pd_mat)) ]
design <- model.matrix(~0+factor(group_list))
colnames(design)=levels(factor(group_list))
rownames(design)=colnames(exprSet)
design
dge <- DGEList(counts=exprSet)
dge <- calcNormFactors(dge)
logCPM <- cpm(dge, log=TRUE, prior.count=3)
v <- voom(dge,design,plot=TRUE, normalize="quantile")
fit <- lmFit(v, design)
con='high-low'
cont.matrix=makeContrasts(contrasts=c(con),
levels = design)
fit2=contrasts.fit(fit,cont.matrix)
fit2=eBayes(fit2)
tempOutput = topTable(fit2, coef=con, n=Inf)
DEG_limma_voom = na.omit(tempOutput)
head(DEG_limma_voom)
return(DEG_limma_voom)
})
得到的 stromal_DEG_limma_voom_list 变量里面存储着全部的癌症内部自己的StromalScore高低分组后的差异分析结果, 而且都是走limma的voom算法的差异分析,每个癌症的差异分析都会得到一个结果表格,类似如下:
> head(stromal_DEG_limma_voom_list[[1]])
logFC AveExpr t P.Value adj.P.Val B
IGFBP4 1.995583 6.5035798 8.183934 3.223932e-12 5.277577e-08 17.47925
ARHGAP15 1.403659 1.6762521 7.531687 6.186283e-11 5.063473e-07 14.54074
PTGER4 2.006874 0.5003204 7.435931 9.520640e-11 5.195096e-07 13.97941
LPAR6 1.470171 3.5192106 7.054566 5.251671e-10 2.149246e-06 12.58534
RASSF2 1.351020 2.7372996 6.906117 1.016025e-09 2.380552e-06 11.95714
ENG 1.067536 6.5360596 6.929772 9.147926e-10 2.380552e-06 11.94358
前面的代码简单修改,就可以得到 Immune_DEG_limma_voom_list 变量里面存储着全部的癌症内部自己的 ImmuneScore 高低分组后的差异分析结果!
Immune_DEG_limma_voom_list <- lapply(tp, function(i){
# i=tp[1]
tp_scores=cg_scores[cg_scores$type==i,]
group_list=ifelse(tp_scores$ImmuneScore > median(tp_scores$ImmuneScore ),'high','low')
exprSet=pd_mat[,match(gsub('[.]','-',rownames(tp_scores)),
colnames(pd_mat)) ]
design <- model.matrix(~0+factor(group_list))
colnames(design)=levels(factor(group_list))
rownames(design)=colnames(exprSet)
design
dge <- DGEList(counts=exprSet)
dge <- calcNormFactors(dge)
logCPM <- cpm(dge, log=TRUE, prior.count=3)
v <- voom(dge,design,plot=TRUE, normalize="quantile")
fit <- lmFit(v, design)
con='high-low'
cont.matrix=makeContrasts(contrasts=c(con),
levels = design)
fit2=contrasts.fit(fit,cont.matrix)
fit2=eBayes(fit2)
tempOutput = topTable(fit2, coef=con, n=Inf)
DEG_limma_voom = na.omit(tempOutput)
head(DEG_limma_voom)
return(DEG_limma_voom)
})
> tp
[1] "ACC" "BLCA" "BRCA" "CESC" "CHOL" "COAD" "DLBC" "ESCA" "GBM" "HNSC" "KICH"
[12] "KIRC" "KIRP" "LAML" "LGG" "LIHC" "LUAD" "LUSC" "MESO" "OV" "PAAD" "PCPG"
[23] "PRAD" "READ" "SARC" "SKCM" "STAD" "TGCT" "THCA" "THYM" "UCEC" "UCS" "UVM"
names(stromal_DEG_limma_voom_list)=tp
names(Immune_DEG_limma_voom_list)=tp
save(stromal_DEG_limma_voom_list,Immune_DEG_limma_voom_list,
file='DEG_limma_voom_list_by_estimate_in_all_tcga_cancers.Rdata')
多次差异分析上下调基因数量结果可视化
代码如下所示:
logFC_cut = 1
padj_cut = 0.05
stat = lapply( stromal_DEG_limma_voom_list , function(DEG){
DEG_sig = subset(DEG, abs(logFC)> logFC_cut &
adj.P.Val< padj_cut)
tb=table(DEG_sig$logFC>0)
up_total = as.numeric(tb["TRUE"])
down_total = as.numeric(tb["FALSE"])
return(c(up_total, down_total))
})
stat = as.data.frame(do.call(rbind,stat))
colnames(stat) = c("UP","DOWN")
rownames(stat) = names(stromal_DEG_limma_voom_list)
stat$Type = rownames(stat)
stat
# stat_reshaped=reshape2::melt(stat)
# head(stat_reshaped)
stat_up = subset(stat, select = -DOWN)
stat_up$group = "UP"
stat_down = subset(stat, select = -UP)
stat_down$label = stat_down$DOWN
stat_down$DOWN = -1*stat_down$DOWN
stat_down$group = "DOWN"
p_multi_DEG=ggplot() +
geom_bar(data = stat_up, aes(x=Type, y=UP, fill=group),stat = "identity",position = 'dodge') +
geom_text(data = stat_up, aes(x=Type, y=UP, label=UP, vjust=-0.25)) +
geom_bar(data = stat_down, aes(x=Type, y=DOWN, fill=group),stat = "identity",position = 'dodge')+
geom_text(data = stat_down, aes(x=Type, y=DOWN, label=label, vjust= 1.25)) +
scale_fill_manual(values=c("#0072B5","#BC3C28")) +
theme(axis.text.x = element_text(angle = 45, vjust = 0.8, hjust=0.8, size = 12)) +
xlab(label = "") + ylab(label = "DEG numbers") +
ggtitle("logFC cut-off : 1\nPadj cut-off : 0.05")
p_multi_DEG
ggsave(filename = "multi_DEG_stromal_DEG_limma_voom_list.pdf",
plot = p_multi_DEG, width = 8, height = 10)
出图如下所示:
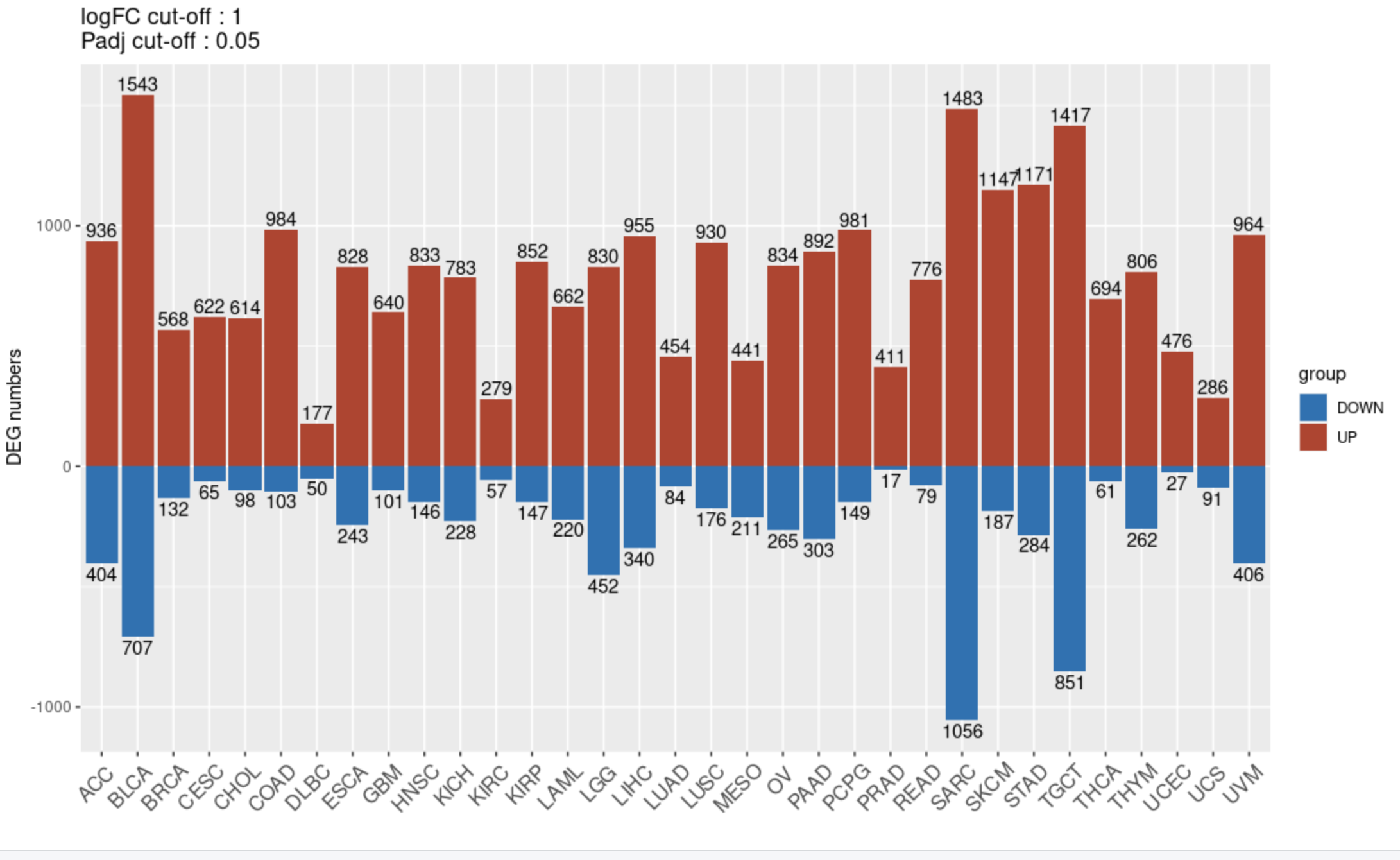
每个癌症内部的StromalScore高低分组后的差异分析结果,上下调基因数量不一样,上面的差异基因数量的条形图背后都是有对应的热图以及火山图。有了基因列表也可以很容易去做生物学功能富集分析。
同理,ImmuneScore高低分组后的差异分析结果也可以同样的可视化!
多次差异分析结果gsea富集分析可视化
首先整合全部的差异分析结果成为矩阵:
# 基因的上下调排序,进行批量ssGSEA分析
# 这里仅仅是展现 stromal_DEG_limma_voom_list 的操作
lapply(stromal_DEG_limma_voom_list,dim)
allgenes=rownames(stromal_DEG_limma_voom_list[[1]])
stromal_logFC_df = as.data.frame( do.call(cbind,
lapply(stromal_DEG_limma_voom_list,function(deg){
deg[allgenes,1]
})))
rownames(stromal_logFC_df)=allgenes
colnames(stromal_logFC_df)=names(stromal_DEG_limma_voom_list)
stromal_logFC_df[1:5,1:5]
> stromal_logFC_df[1:5,1:5]
ACC BLCA BRCA CESC CHOL
IGFBP4 1.995583 0.61560996 0.7019134 0.5863464 0.7060882
ARHGAP15 1.403659 1.35184683 0.6921096 1.0444763 1.4553690
PTGER4 2.006874 -0.09279879 0.9179623 0.1816853 1.5790391
LPAR6 1.470171 0.29102849 0.6212784 0.7616387 0.8350077
RASSF2 1.351020 0.55992731 0.9519274 0.9826369 1.3343867
然后针对human的HALLMARK的50个基因集,对全部的33个癌症的差异分析结果,批量做ssGSEA分析,代码如下所示:
# install.packages("msigdbr")
library(msigdbr)
h_gene_sets = msigdbr(species = "human", category = "H")
msigdbr_list = split(x = h_gene_sets$gene_symbol,
f = h_gene_sets$gs_name)
msigdbr_list
library(GSVA)
library(limma)
library(GSEABase)
library(data.table)
msigdbr_list
# 参考:http://www.bio-info-trainee.com/6602.html
gsc <- GeneSetCollection(mapply(function(geneIds, keggId) {
GeneSet(unique(geneIds), geneIdType=EntrezIdentifier(),
collectionType=KEGGCollection(keggId),
setName=keggId)
}, msigdbr_list, names(msigdbr_list)
))
gsc
stromal_ssgseaScore=gsva(as.matrix(stromal_logFC_df), gsc,
method='ssgsea', kcdf='Gaussian',
abs.ranking=TRUE)
rownames(stromal_ssgseaScore)=gsub('HALLMARK_','',rownames(stromal_ssgseaScore))
pheatmap(stromal_ssgseaScore)
出图如下:
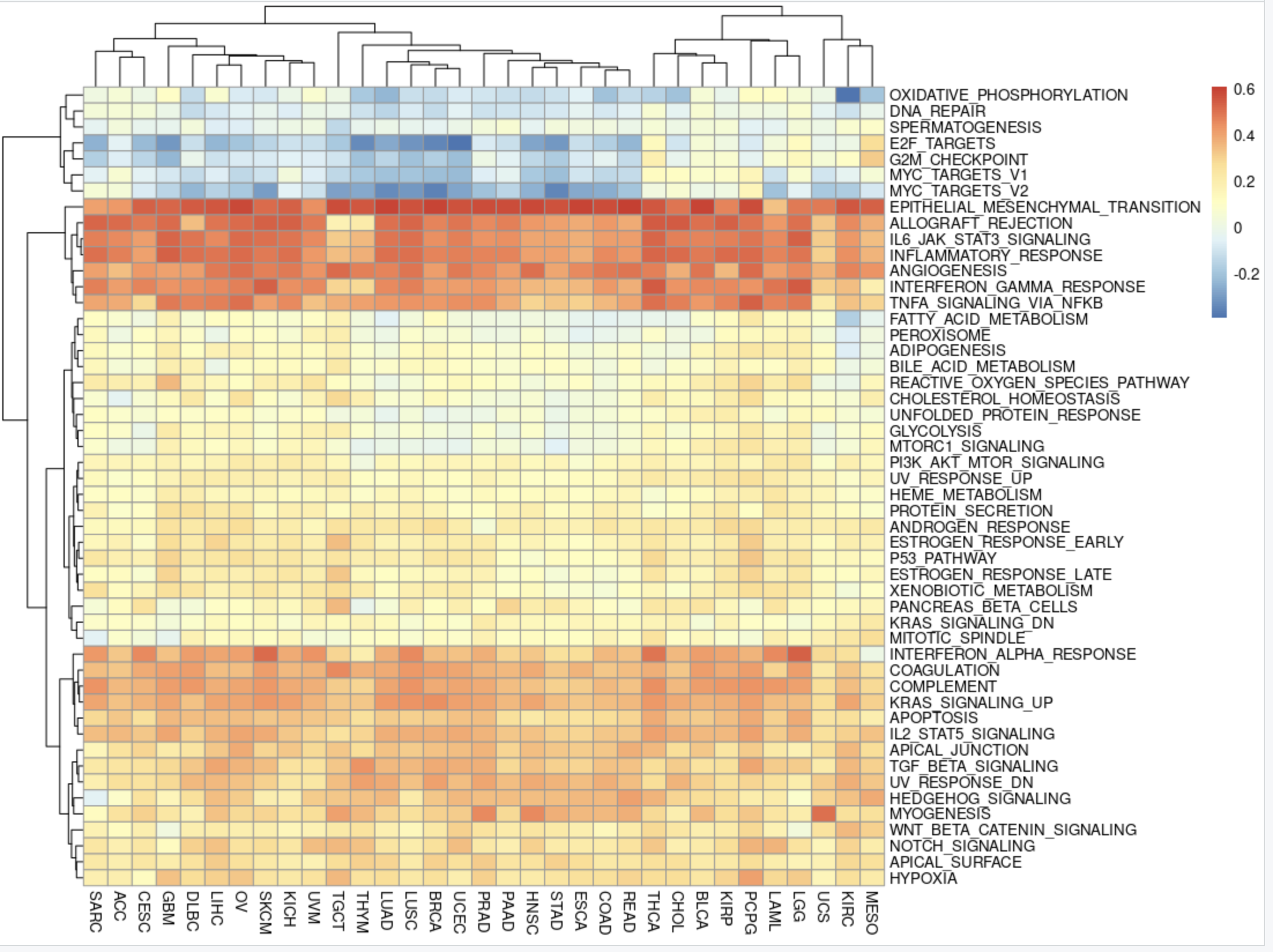
挺有意思的,可以看到不同癌症内部按照StromalScore高低分组后的差异分析结果的基因排序后,针对human的HALLMARK的50个基因集批量做ssGSEA分析出奇的稳定!
同理,我们可以对ImmuneScore高低分组后的差异分析结果做类似的分析!
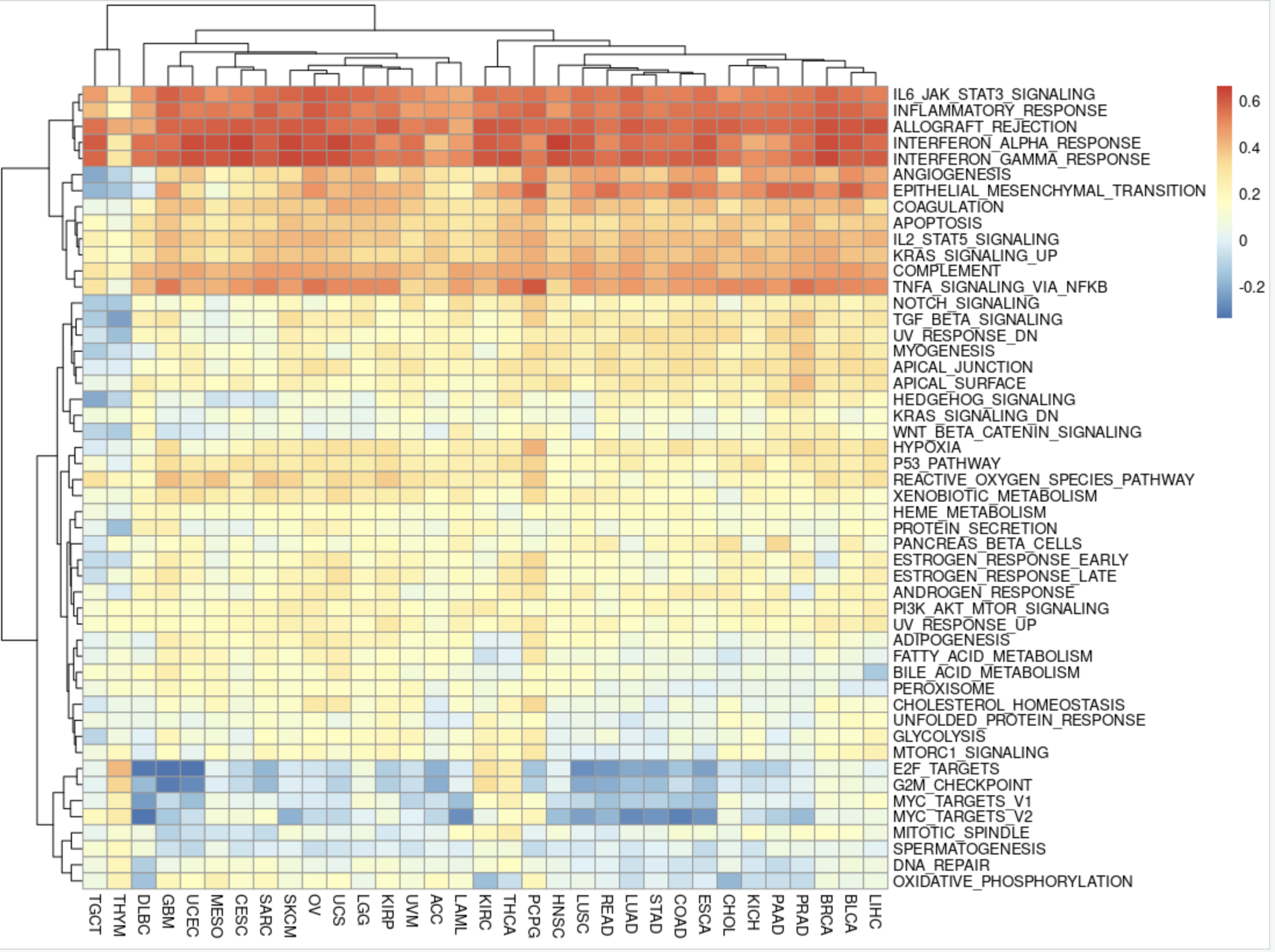
不过 MSigDB(Molecular Signatures Database)数据库太丰富了,我们仅仅是看了看human的HALLMARK的50个基因集。其实该数据库中定义了已知的基因集合:http://software.broadinstitute.org/gsea/msigdb 包括H和C1-C7的 八个系列(Collection),每个系列分别是:
- H: hallmark gene sets (癌症)特征基因集合,共50组,最常用;
- C1: positional gene sets 位置基因集合,根据染色体位置,共326个,用的很少;
- C2: curated gene sets:(专家)校验基因集合,基于通路、文献等:
- C3: motif gene sets:模式基因集合,主要包括microRNA和转录因子靶基因两部分
- C4: computational gene sets:计算基因集合,通过挖掘癌症相关芯片数据定义的基因集合;
- C5: GO gene sets:Gene Ontology 基因本体论,包括BP(生物学过程biological process,细胞原件cellular component和分子功能molecular function三部分)
- C6: oncogenic signatures:癌症特征基因集合,大部分来源于NCBI GEO 发表芯片数据
- C7: immunologic signatures: 免疫相关基因集合。
任意条件都可以在各个癌症内部进行分组然后批量差异和注释
大家已经看到了estimate的两个打分值结果都是可以进行各个癌症内部进行分组然后 差异和注释,那么简单的编程思维就可以告诉你,这样的操作可以批量化!
除了针对estimate的两个打分值结果可以分组,你可以针对缺氧,自噬,免疫等指标在各个癌症内部进行分组然后批量差异和注释,然后比较不同癌症的表现差异。
思考题
大家可以自行根据自己的生物学背景提出一些打分或者说分组的指标,在全部的癌症内部批量分组差异和注释哦!