前面我们公布了《cytof数据资源介绍(文末有交流群)》,现在就开始正式手把手教学。
上一讲我们构造好了SingleCellExperiment对象,后续全部的分析都会以这个SingleCellExperiment对象为准,大家务必熟悉SingleCellExperiment对象的各种结构,教程见:cytofWorkflow之构建SingleCellExperiment对象(二)。有了这个SingleCellExperiment对象,第一步是对它做一些质量控制操作。其实前面教程里面的看这些抗体在我们的不同病人的表达量分布情况:
require(cytofWorkflow)
p <- plotExprs(sce, color_by = "condition")
p$facet$params$ncol <- 6
p
密度图显示,信号值被归一化。这已经是一种质量控制的啦。
再次回顾一下SingleCellExperiment对象构建的代码
全部的可以复制粘贴就执行的代码如下:
require(cytofWorkflow)
if(!file.exists(file = 'input_for_cytofWorkflow.Rdata')){
library(readxl)
url <- "http://imlspenticton.uzh.ch/robinson_lab/cytofWorkflow"
md <- "PBMC8_metadata.xlsx"
download.file(file.path(url, md), destfile = md, mode = "wb")
md <- read_excel(md)
head(data.frame(md))
table(md[,3:4])
# 样本的表型信息
## 真正的表达矩阵
library(HDCytoData)
fs <- Bodenmiller_BCR_XL_flowSet()
fs
# 如果网络不好,也可以自行下载
# 然后:loaded into R as a flowSet using read.flowSet() from the flowCore package
# 这个cytof的panel的抗体信息表格:
panel <- "PBMC8_panel_v3.xlsx"
download.file(file.path(url, panel), destfile = panel, mode = "wb")
panel <- read_excel(panel)
head(data.frame(panel))
# spot check that all panel columns are in the flowSet object
all(panel$fcs_colname %in% colnames(fs))
# 有了样本的表型信息,panel的抗体信息,以及表达量矩阵,就可以构建对象:
# specify levels for conditions & sample IDs to assure desired ordering
md$condition <- factor(md$condition, levels = c("Ref", "BCRXL"))
md$sample_id <- factor(md$sample_id,
levels = md$sample_id[order(md$condition)])
save(fs, panel, md,file = 'input_for_cytofWorkflow.Rdata')
}
load(file = 'input_for_cytofWorkflow.Rdata')
# expression values, 第一个样品是2838个细胞,39列(其中6列并不是抗体)
dim(exprs(fs[[1]]))
exprs(fs[[1]])[1:6, 1:5]
colnames(exprs(fs[[1]]))
# construct SingleCellExperiment
panel
md
library(CATALYST)
sce <- prepData(fs, panel, md, features = panel$fcs_colname)
pro='basic_qc'
p <- plotExprs(sce, color_by = "condition")
p$facet$params$ncol <- 4
p
ggsave2(filename = paste0(pro,'_all_markers_density.pdf'))
第一个图如下所示:
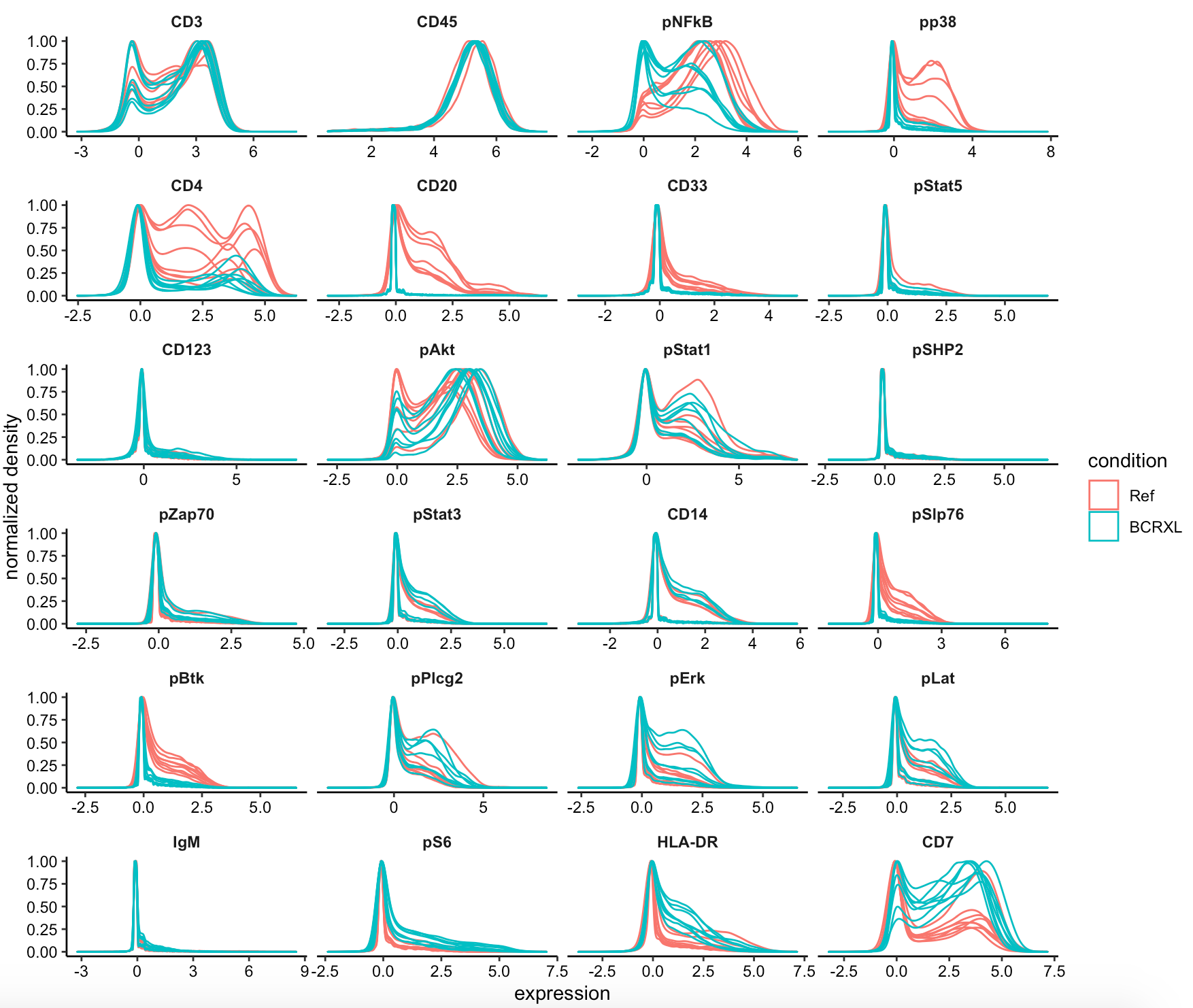
可以看到,某些抗体在两个不同组的表达量差异很明显, 他们将会是主要的细胞亚群差异来源。
然后看看2个分组的各自8个样品的细胞数量
代码也超级简单,这就是构建SingleCellExperiment对象的方便之处,如下:
n_cells(sce)
plotCounts(sce, group_by = "sample_id", color_by = "condition")
ggsave2(filename = paste0(pro,'_plotCounts.pdf'))
可以看到细胞数量差异是比较大的,这个是cytof数据的客观规律,不用过于在意。
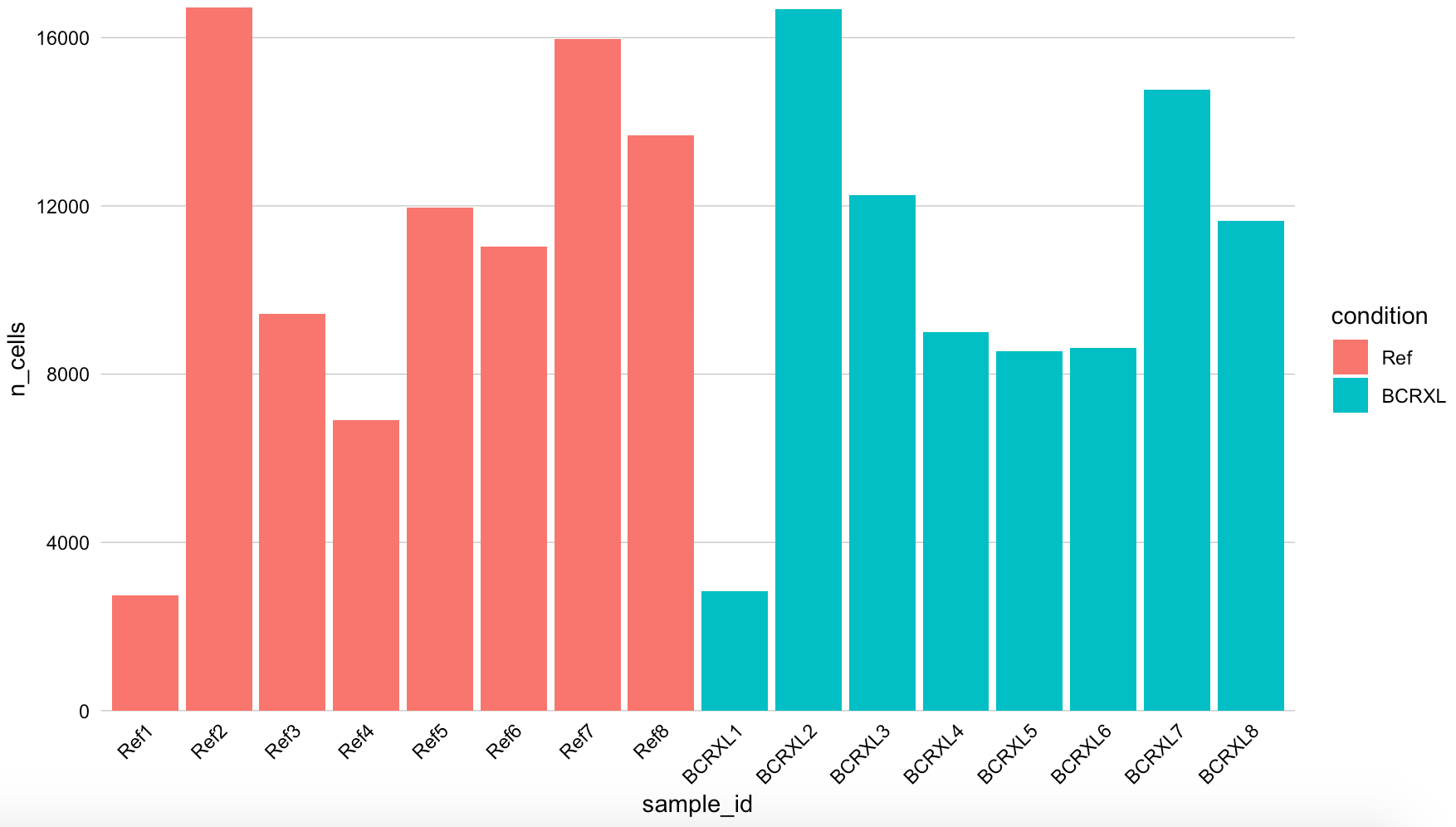
当然了,也可以自己写代码去看细胞数量,然后自己绘制这个条形图:
df=data.frame(n_cells=unlist(lapply(1:length(fs), function(i){
dim(exprs(fs[[i]]))[1]
})),
condition=md$condition,sample_id=md$sample_id)
library(ggplot2)
ggplot2::ggplot(df,aes(x=sample_id,y=n_cells,fill=condition))+ geom_bar(stat = "identity")
当然了,需要调整很多细节啦,其实看看作者自己的 plotCounts 函数就明白了。绝大部分情况下,初学者都无需自己写太多函数,各种工具调用的溜起来就行了。
然后看看样品之间的距离关系
代码也超级简单,这就是构建SingleCellExperiment对象的方便之处,如下:
# pbMDS: Pseudobulk-level MDS plot.
# A multi-dimensional scaling (MDS) plot on aggregated measurement values may be rendered with pbMDS.
pbMDS(sce, color_by = "condition", label_by = "sample_id")
ggsave2(filename = paste0(pro,'_pbMDS.pdf'))
可以看到cytofWorkflow给出来的示例数据超级正常,不同分组的病人距离很远,泾渭分明,如下:
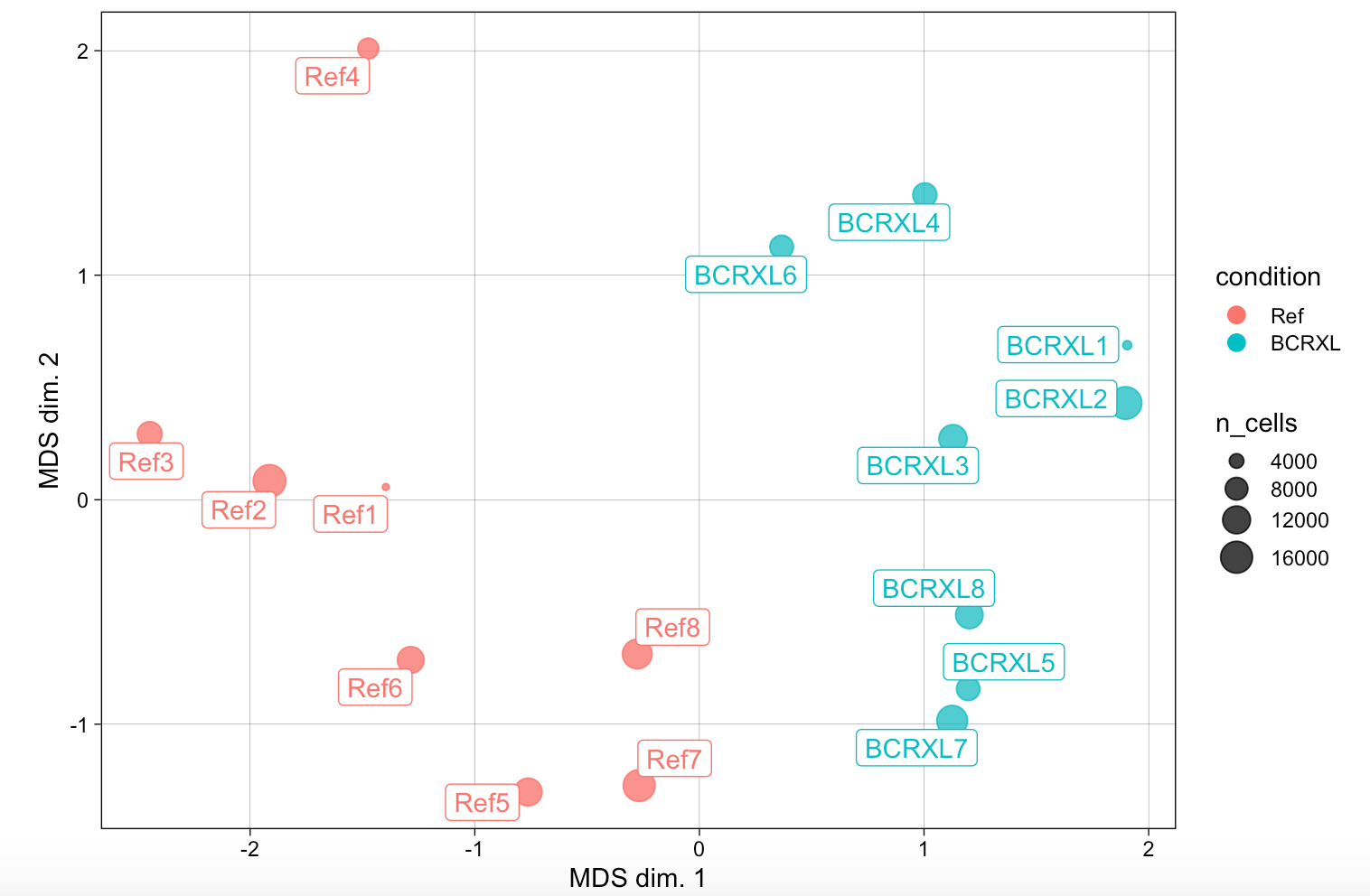
这样的图表在我一直讲解的表达矩阵分析也是如此。很多人仅仅是学习了代码,不明白后面的统计学意义,PCA图的意义,我一直强调,做表达矩阵分析一定要有三张图,见:你确定你的差异基因找对了吗? 。
这里面的MDS看样本距离关系,跟我们一直强调的PCA看样本距离关系,大同小异。
表达量热图看差异
代码也超级简单,这就是构建SingleCellExperiment对象的方便之处,如下:
# 10 lineage markers and 14 functional markers across all cells measured for each sample in the PBMC dataset
pdf( paste0(pro,'_plotExprHeatmap.pdf'))
plotExprHeatmap(sce, scale = "last",
hm_pal = rev(hcl.colors(10, "YlGnBu")))
dev.off()
出图如下:
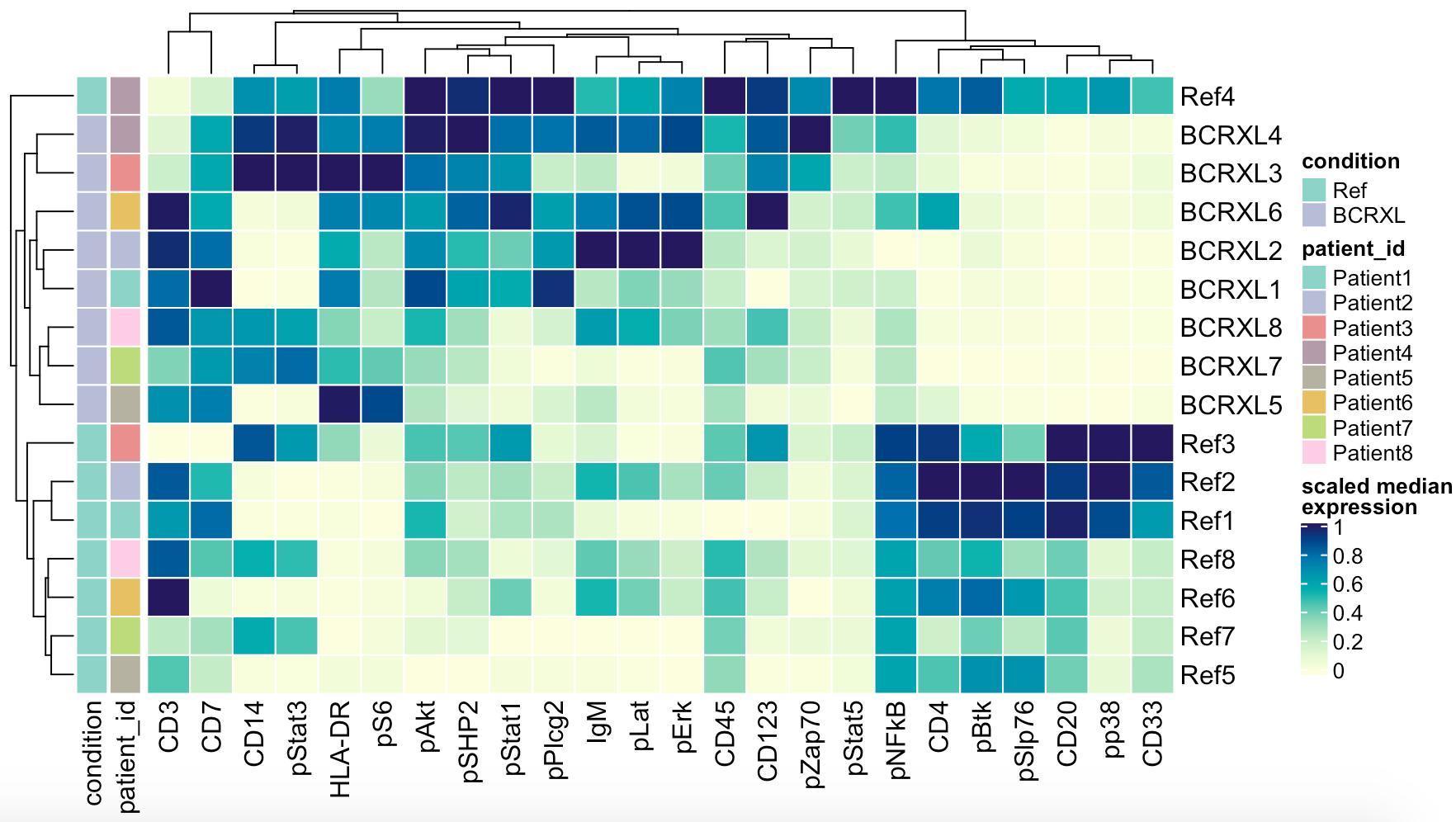
可以看到,在Ref组里面非常多的CD4表达,在BCRXL组却低,所以最后对这些样本的细胞进行聚类分群后,是可以看到CD4阳性T细胞的比例差异的。
另外一种展现表达量差异的条形图
代码也超级简单,这就是构建SingleCellExperiment对象的方便之处,如下:
# PCA-based non-redundancy score (NRS)
plotNRS(sce, features = "type", color_by = "condition")
ggsave2(filename = paste0(pro,'_plotNRS.pdf'))
出图如下:
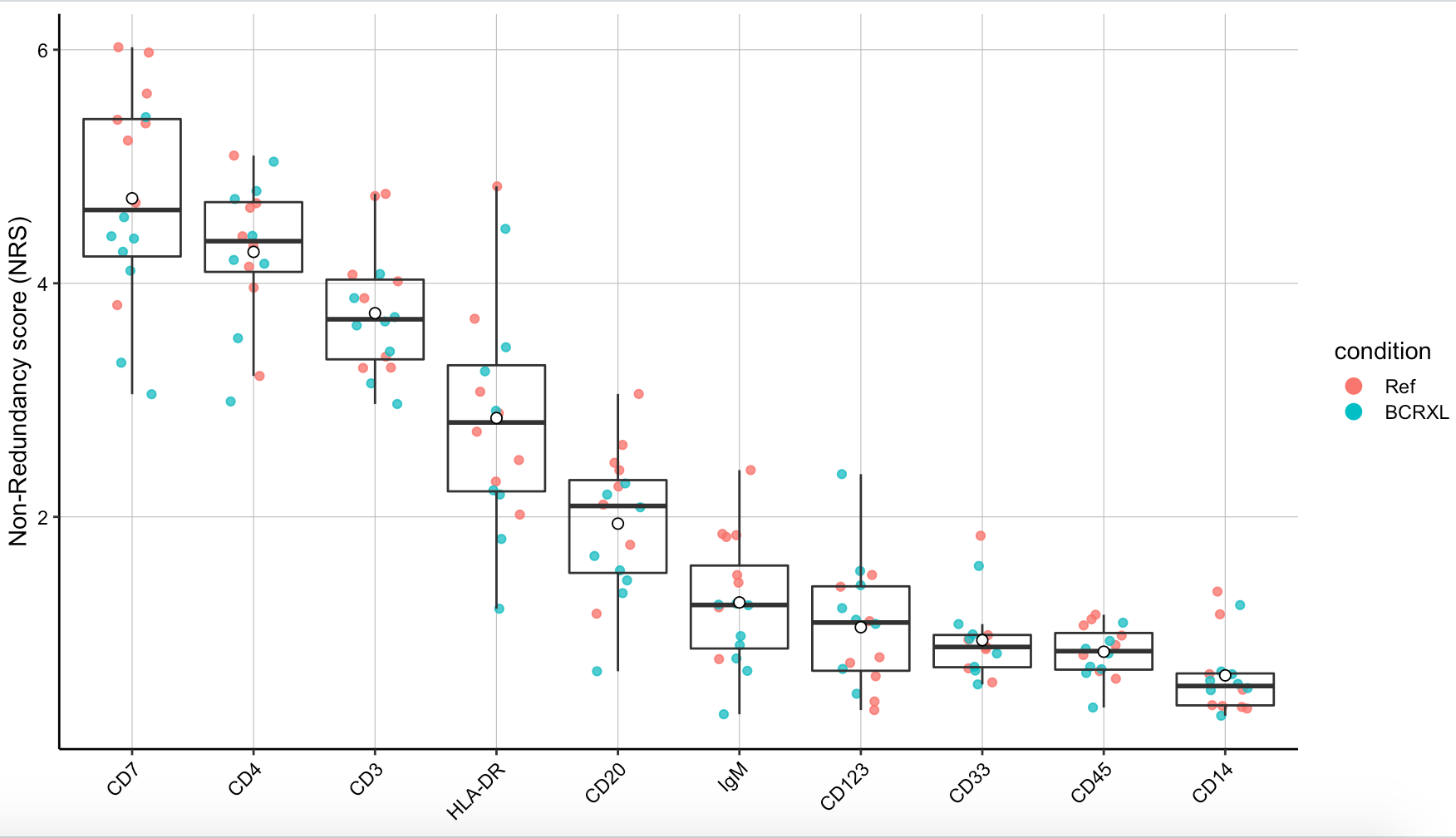
PCA-based non-redundancy score 比较难理解,反正绝大部分情况下我是忽略这张图的,虽然说cytofWorkflow教程里面展现了它。
其实这个时候的分析,跟纯粹的单细胞转录组就非常类似了。单细胞转录组数据分析的细节,以及背景我就不赘述了,看我在《单细胞天地》的单细胞基础10讲:
- 01. 上游分析流程
- 02.课题多少个样品,测序数据量如何
- 03. 过滤不合格细胞和基因(数据质控很重要)
- 04. 过滤线粒体核糖体基因
- 05. 去除细胞效应和基因效应
- 06.单细胞转录组数据的降维聚类分群
- 07.单细胞转录组数据处理之细胞亚群注释
- 08.把拿到的亚群进行更细致的分群
- 09.单细胞转录组数据处理之细胞亚群比例比较
以及各式各样的个性化汇总教程,差不多就明白了。如果你们有单细胞转录组数据处理经验其实就很容易理解cytof数据分析啦。值得注意的是cytof数据呢,直接机器下机就是FCS文件,可以R语言无缝连接的读取继续下游分析。但是单细胞转录组以10X公司为主流,我们也是在单细胞天地公众号详细介绍了cellranger流程,大家可以自行前往学习,如下:
- 单细胞实战(一)数据下载
- 单细胞实战(二) cell ranger使用前注意事项
- 单细胞实战(三) Cell Ranger使用初探
- 单细胞实战(四) Cell Ranger流程概览
- 单细胞实战(五) 理解cellranger count的结果
但是这个两年前的系列笔记是基于V2,V3版本的cellranger,目前呢它更新到了版本4,建议以我的最新版教程为准,在《生信技能树》的教程:cellranger更新到4啦(全新使用教程)