前面我们详细讲解过,sanger研究所科学家【1】提出来了肿瘤somatic突变的signature概念 ,把96突变频谱的非负矩阵分解后的30个特征,在cosmic数据库可以学习它。不同的特征有不同的生物学含义【2】,比如文章【3】 就是使用了 这些signature区分生存!主要是R包deconstructSigs可以把自己的96突变频谱对应到cosmic数据库的30个突变特征。
- 【1】https://software.broadinstitute.org/cancer/cga/msp
- 【2】https://en.wikipedia.org/wiki/Mutational_signatures
- 【3】https://www.nature.com/articles/s41586-019-1056-z
另外一个策略就是自己推断denovo的signature,可以使用SomaticSignatures 包的identifySignatures函数。这个教程我也在生信技能树分享过:使用R包SomaticSignatures进行denovo的signature推断
而且前面我在生信菜鸟团的肿瘤外显子数据分析专辑提到了,很多研究者会嫌弃cosmic数据库的30个肿瘤突变signatures,他们觉得cosmic数据库30个signature的生物学意义并不好,会尝试自己分解出来自己的signature。比如:0元,10小时教学视频直播《跟着百度李彦宏学习肿瘤基因组测序数据分析》 这个文献,研究者就是使用R包SomaticSignatures进行denovo的signature推断,拿到了11个自定义的signature。
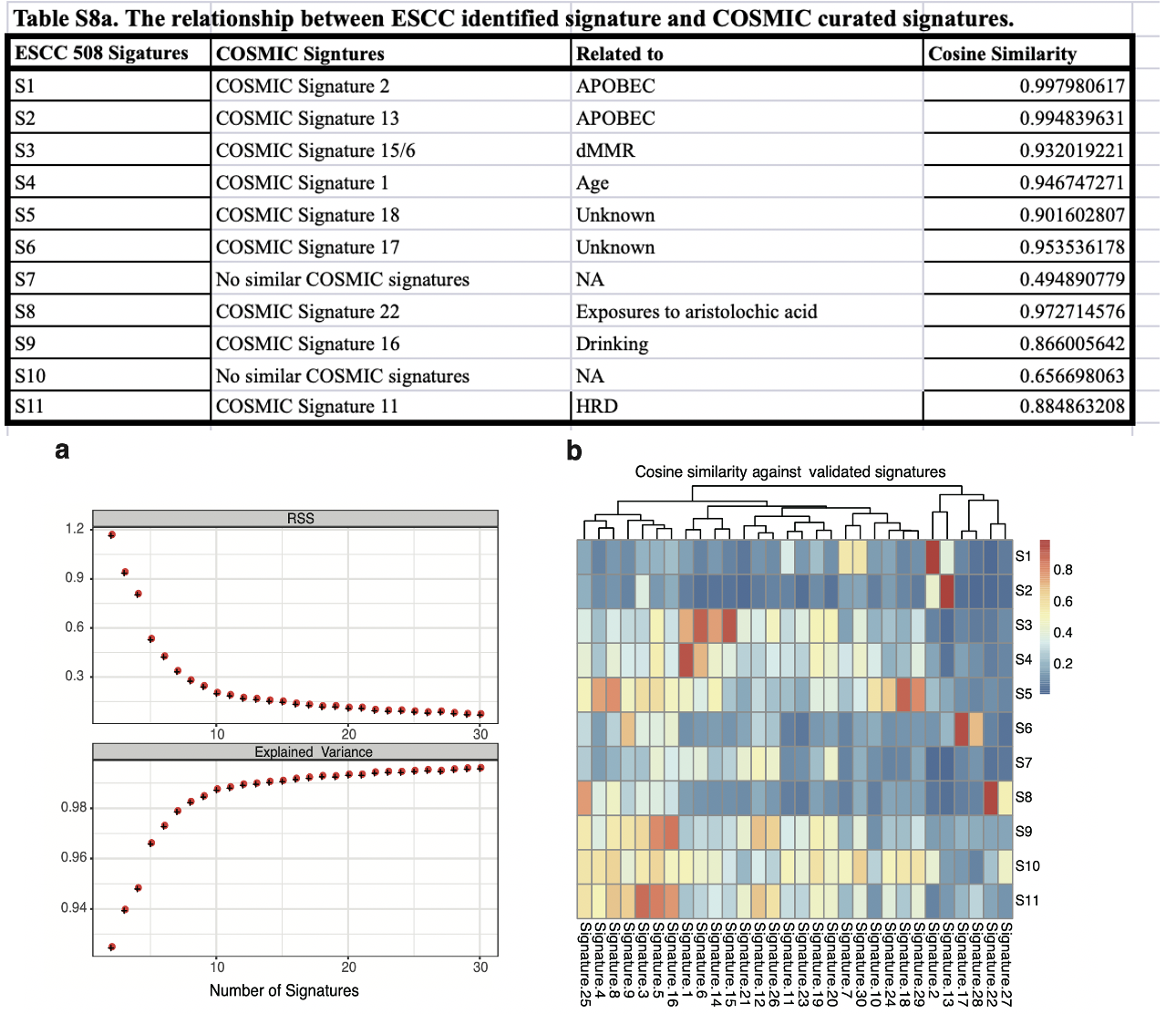
这个时候,通常的分析节奏,就是把11个自定义的signature去和cosmic数据库的30个突变特征进行对比。原文图表如下:
signatures的本质就是96突变形式的比例,所以可以直接在R里面进行相关性计算。
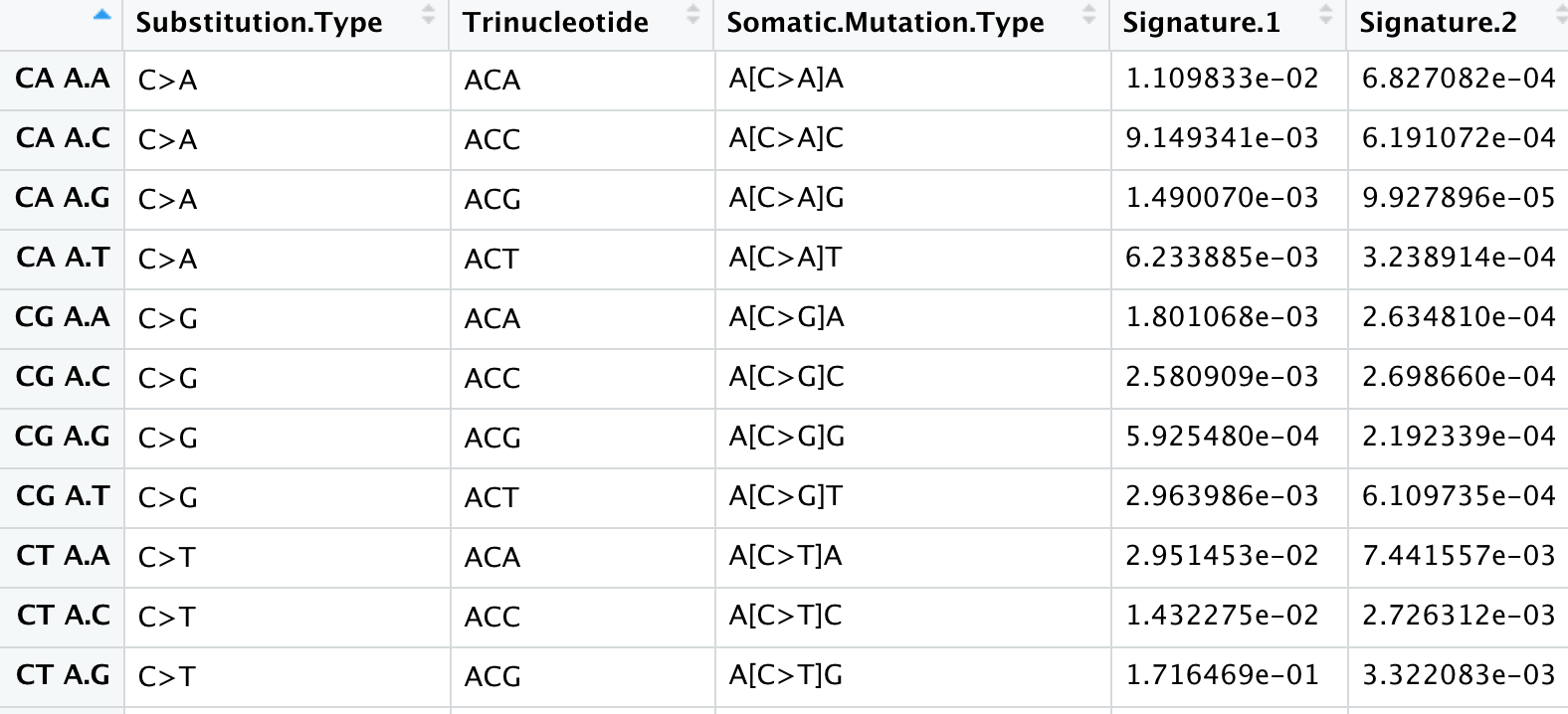
首先查看自己的11个自定义的signature的96突变频谱
下面的escc_denovo_results.Rata文件来源于前面的教程:
load(file = 'escc_denovo_results.Rata')
str(sigs_nmf)
# sp signatures_probabilities
sp=sigs_nmf@signatures
head(sp)
colSums(sp)
sp=apply(sp,2,function(x){
x/sum(x)
})
denovo=sp
rownames(denovo)
可以看到,自己的11个自定义的signature的96突变频谱如下:

然后查看cosmic的30个signature的96突变频谱
需要读取网络文件,signatures_probabilities.txt,代码如下:
# https://cancer.sanger.ac.uk/cancergenome/assets/signatures_probabilities.txt
cosmic=read.table('https://cancer.sanger.ac.uk/cancergenome/assets/signatures_probabilities.txt',
header = T,sep = '\t')[,1:33]
head(cosmic[,1:3])
tmp=cosmic[,2];substr(tmp,2,2) <- '.'
rownames(cosmic)=paste(gsub('>','',cosmic[,1]),
tmp)
简单的转换,保证两个signature的矩阵行名是一样的
绘制相关性热图
代码如下:
comp=cbind(denovo[rownames(cosmic),],
cosmic[,4:33])
colSums(comp)
pheatmap::pheatmap(cor(comp))
pheatmap::pheatmap(cor(comp)[1:11,12:41])
可以看到:
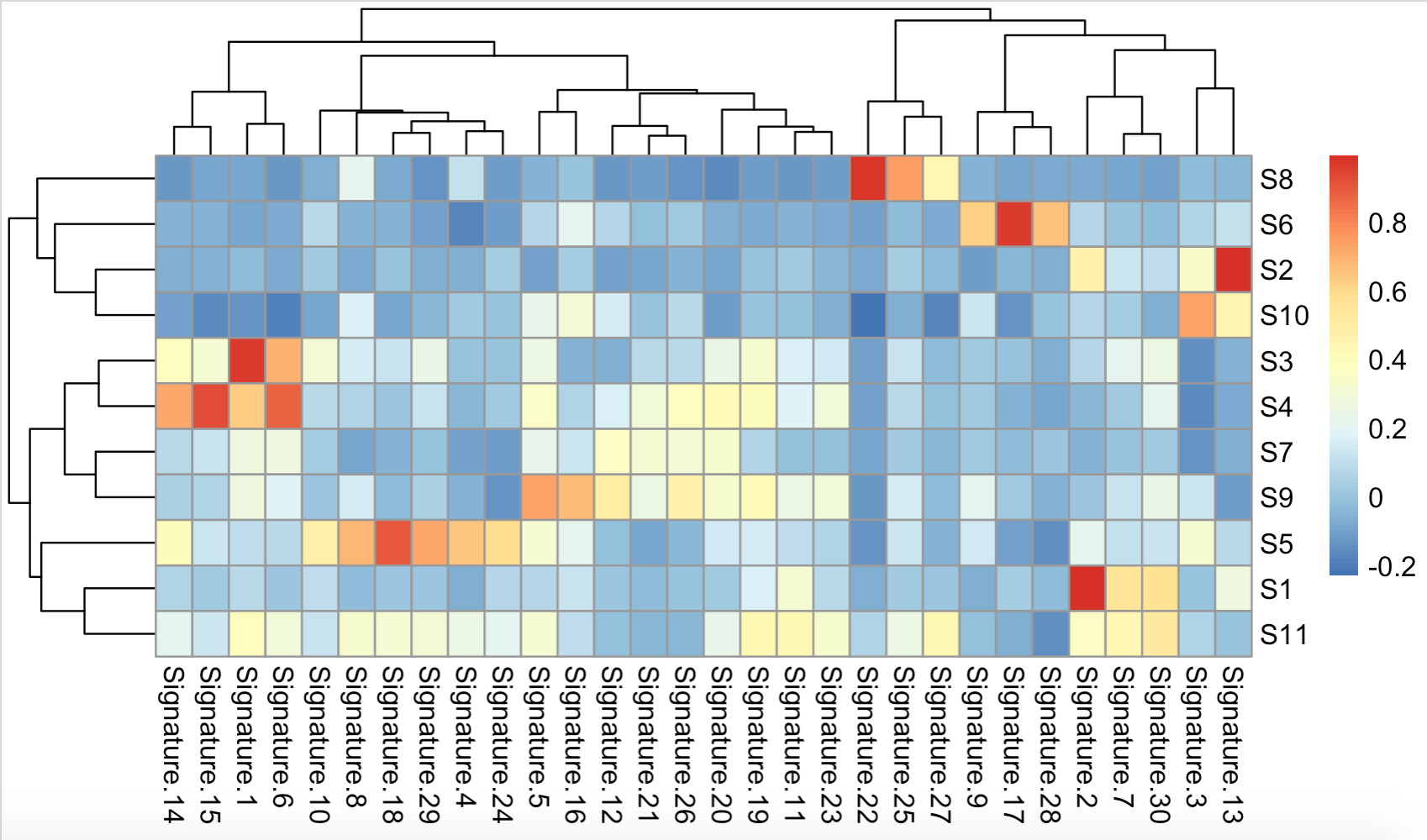
文章里面的不同体系的signature的关系,得到了验证;
ESCC 508 Sigatures COSMIC Signtures
S1 COSMIC Signature 2
S2 COSMIC Signature 13
S3 COSMIC Signature 15/6
S4 COSMIC Signature 1
S5 COSMIC Signature 18
S6 COSMIC Signature 17
S7 No similar COSMIC signatures
S8 COSMIC Signature 22
S9 COSMIC Signature 16
S10 No similar COSMIC signatures
S11 COSMIC Signature 11
你可以去跟Whole-genome sequencing of 508 patients identifies key molecular features associated with poor prognosis in esophageal squamous cell carcinoma文章对比一下,几乎是一模一样。
后面我们还需要针对signature结果,进行NMF聚类后对病人进行分组,计算临床预后意义,欢迎继续follow我们的0元,10小时教学视频直播《跟着百度李彦宏学习肿瘤基因组测序数据分析》 。