我在单细胞天地教程:表达矩阵逆转为10X的标准输出3个文件,详细介绍过 10X文件的3个标准文件:
比如SRR7722939数据集里面,文件barcodes.tsv 和 genes.tsv,就是表达矩阵的行名和列名:
jmzengdeMacBook-Pro:SRR7722939 jmzeng$ head barcodes.tsv
AAACCTGAGCGAAGGG-1
AAACCTGAGGTCATCT-1
AAACCTGAGTCCTCCT-1
AAACCTGCACCAGCAC-1
AAACCTGGTAACGTTC-1
AAACCTGGTAAGGATT-1
AAACCTGGTTGTCGCG-1
AAACCTGTCCTGCCAT-1
AAACGGGAGTCATCCA-1
AAACGGGCATGGATGG-1
jmzengdeMacBook-Pro:SRR7722939 jmzeng$ head genes.tsv
hg38_ENSG00000243485 hg38_RP11-34P13.3
hg38_ENSG00000237613 hg38_FAM138A
hg38_ENSG00000186092 hg38_OR4F5
hg38_ENSG00000238009 hg38_RP11-34P13.7
hg38_ENSG00000239945 hg38_RP11-34P13.8
hg38_ENSG00000239906 hg38_RP11-34P13.14
hg38_ENSG00000241599 hg38_RP11-34P13.9
hg38_ENSG00000279928 hg38_FO538757.3
hg38_ENSG00000279457 hg38_FO538757.2
hg38_ENSG00000228463 hg38_AP006222.2
但是matrix.mtx,就稍微复杂一点,仔细看:
jmzengdeMacBook-Pro:SRR7722939 jmzeng$ head matrix.mtx
%%MatrixMarket matrix coordinate integer general
%
33694 2049 1878957
28 1 1
55 1 2
59 1 1
60 1 1
62 1 1
78 1 2
111 1 1
如果你关注这3个文件的行数:
2049 barcodes.tsv
33694 genes.tsv
1878960 matrix.mtx
就会发现,matrix.mtx文件里面的33694、2049、1878957数值,分别是细胞数量,基因数量,以及有表达量的值的数量。
每个10X样本都是走流程拿到10x单细胞转录组数据的3个文件的表达矩阵,比如数据集 GSE128033 和 GSE135893,你去GEO就可以看到并且下载下面的文件:
2.2M Mar 8 2019 GSM3660655_SC94IPFUP_barcodes.tsv.gz
259K Mar 8 2019 GSM3660655_SC94IPFUP_genes.tsv.gz
26M Mar 8 2019 GSM3660655_SC94IPFUP_matrix.mtx.gz
2.2M Mar 8 2019 GSM3660656_SC95IPFLOW_barcodes.tsv.gz
259K Mar 8 2019 GSM3660656_SC95IPFLOW_genes.tsv.gz
31M Mar 8 2019 GSM3660656_SC95IPFLOW_matrix.mtx.gz
2.2M Mar 8 2019 GSM3660657_SC153IPFLOW_barcodes.tsv.gz
259K Mar 8 2019 GSM3660657_SC153IPFLOW_genes.tsv.gz
33M Mar 8 2019 GSM3660657_SC153IPFLOW_matrix.mtx.gz
2.2M Mar 8 2019 GSM3660658_SC154IPFUP_barcodes.tsv.gz
259K Mar 8 2019 GSM3660658_SC154IPFUP_genes.tsv.gz
31M Mar 8 2019 GSM3660658_SC154IPFUP_matrix.mtx.gz
下游处理的时候,一定要保证这3个文件同时存在,而且在同一个文件夹下面,每一个样本都是3个文件,每一个样本都是同样的代码处理。
示例代码是:
rm(list=ls())
options(stringsAsFactors = F)
library(Seurat)
sce1 <- CreateSeuratObject(Read10X('../10x-results/WT/'),
"wt")
重点就是 Read10X 函数读取文件夹路径,比如:../10x-results/WT/ ,保证文件夹下面有3个文件。每个样本读入R后都有一个seurat对象,就需要合并,那个我以前也在单细胞天地讲解过:
但是最近接受到粉丝提问:在数据集https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE127465下载了下面的3个文件:
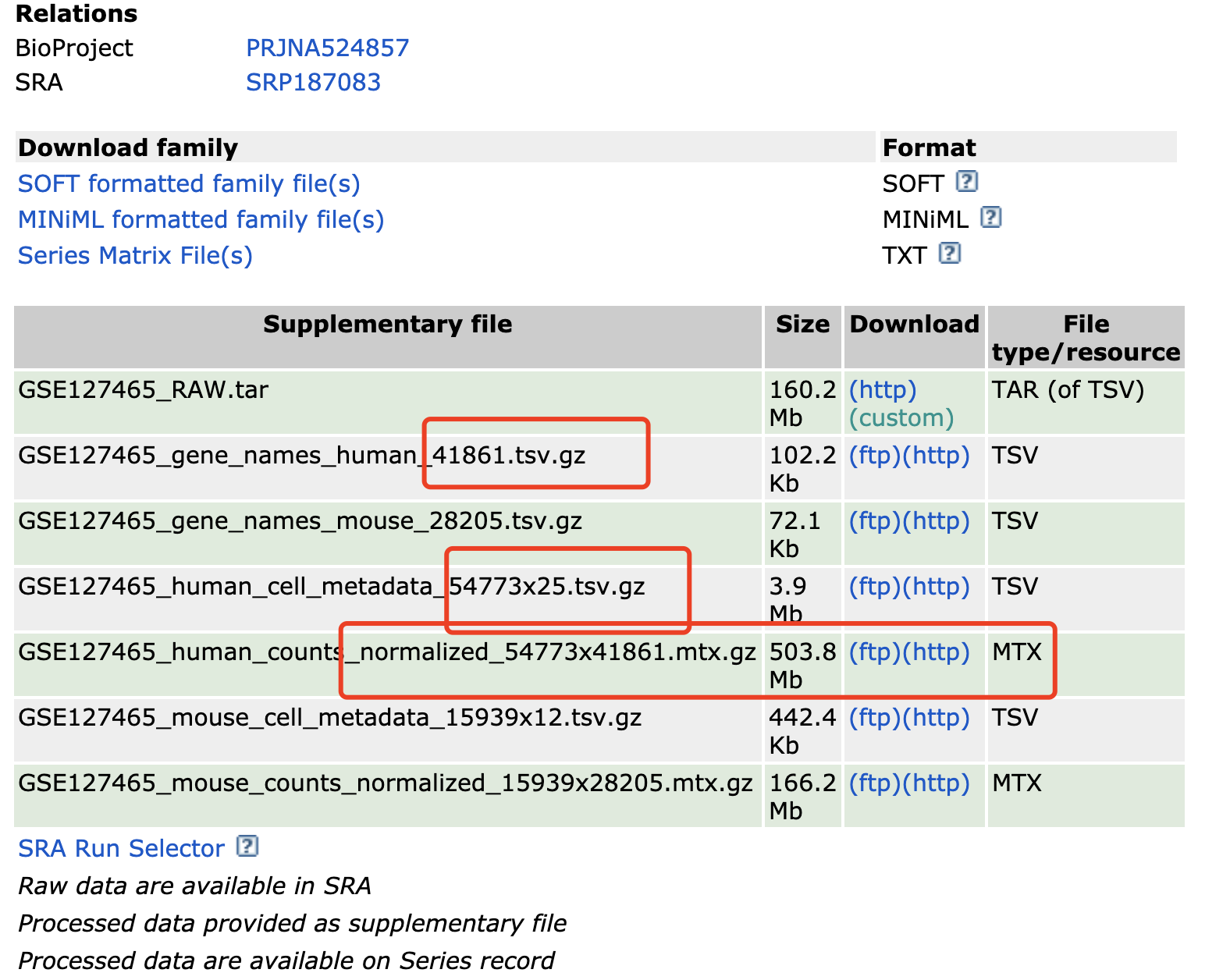
但是却没办法理解它,的确是3个文件,一个mtx.gz ,稀疏矩阵,存储单细胞转录组的表达量,一个是基因信息,一个是细胞信息。
这里先卖一个关子!
明天我们在单细胞天地揭晓答案哈!