有意思的是,如果能合理的提问,绝大部分问题其实就可以自己抽丝剥茧的解决掉
最近有一个学徒非常执着的要自费私聊提问(主要是专家咨询费),这里需要说明一下,我其实每天都会处理十几个粉丝提问,部分比较耗费时间的问题就会让大家等等,因为我也不是每天没事只做公益。除非是实在是等不及的,才需要自费紧急求助于我。
我跟学徒的沟通邮件列表如下:

因为学徒的无效提问,最后我直接让她把代码和数据传递给我了。
错误的代码是
我看了看她错误的代码,主要是参考我表观教程来的,但是她感兴趣的物种是斑马鱼,我的教程是人类研究:
library(rtracklayer)
library(BiocManager)
library(TxDb.Drerio.UCSC.danRer10.refGene)
library(org.Dr.eg.db)
library(ChIPseeker)
narrowPeaksFile = 'wt_peaks.narrowPeak';
narrowPeaksFile
txdb <- TxDb.Drerio.UCSC.danRer10.refGene
library(clusterProfiler)
peak <- readPeakFile(narrowPeaksFile)
peak1<-import(narrowPeaksFile)
keepChr= !grepl('_',seqlevels(peak))
keepChr
seqlevels(peak, pruning.mode="coarse") <- seqlevels(peak)[keepChr]
peakAnno <- annotatePeak(peak, tssRegion=c(-3000, 3000),
TxDb=txdb, annoDb="org.Dr.eg.db")
她还算是知道要替换相关的R包,比如org.Dr.eg.db和TxDb.Drerio.UCSC.danRer10.refGene,其实也是咨询了我,见:
这个代码报错如下:
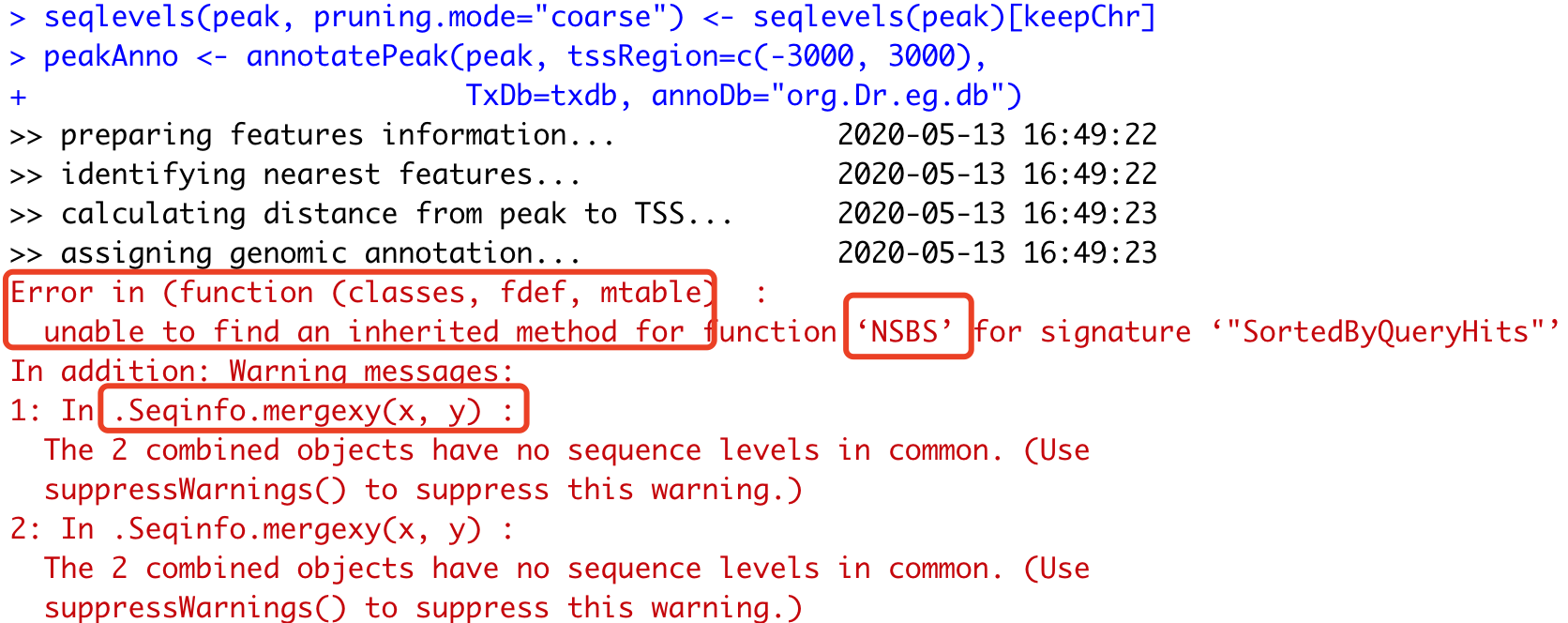
这也就是她为什么反复强调,前面的运行结果也没有报错。但是不知道为什么最后一步就错了。如果没有太多R经验,的确会容易陷入报错细节里面去。
我修改后的代码
其实我一眼也看不出问题所在,因为这个R包也不是我写的,但是我跟她不一样的地方在于,我并不会认为这个错误的产生仅仅是因为最后一个代码,所以我把前面的代码,每个都仔细查看了,修改后的代码是:
rm(list = ls())
library(rtracklayer)
library(BiocManager)
library(TxDb.Drerio.UCSC.danRer10.refGene)
library(org.Dr.eg.db)
library(ChIPseeker)
narrowPeaksFile = 'wt_peaks.narrowPeak';
narrowPeaksFile
txdb <- TxDb.Drerio.UCSC.danRer10.refGene
txdb
head(seqlevels(txdb))
library(clusterProfiler)
peak <- readPeakFile(narrowPeaksFile)
peak
peak1<-import(narrowPeaksFile)
seqlevels(peak)
keepChr= !grepl('K',seqlevels(peak))
keepChr
seqlevels(peak, pruning.mode="coarse") <- seqlevels(peak)[keepChr]
seqlevels(peak)
seqlevels(peak)=paste0('chr',seqlevels(peak))
peak[1:10]
peakAnno <- annotatePeak(peak, tssRegion=c(-3000, 3000),
TxDb=txdb, annoDb="org.Dr.eg.db")
可以看到,代码增加了很多,主要是不停的查看一些变量情况,结果我就发现了,因为她前面走表观流程拿到的wt_peaks.narrowPeak的参考基因组里面是没有chr的,但是两个包org.Dr.eg.db和TxDb.Drerio.UCSC.danRer10.refGene是有的,所以我把那个peaks加上了chr就完美解决问题啦。
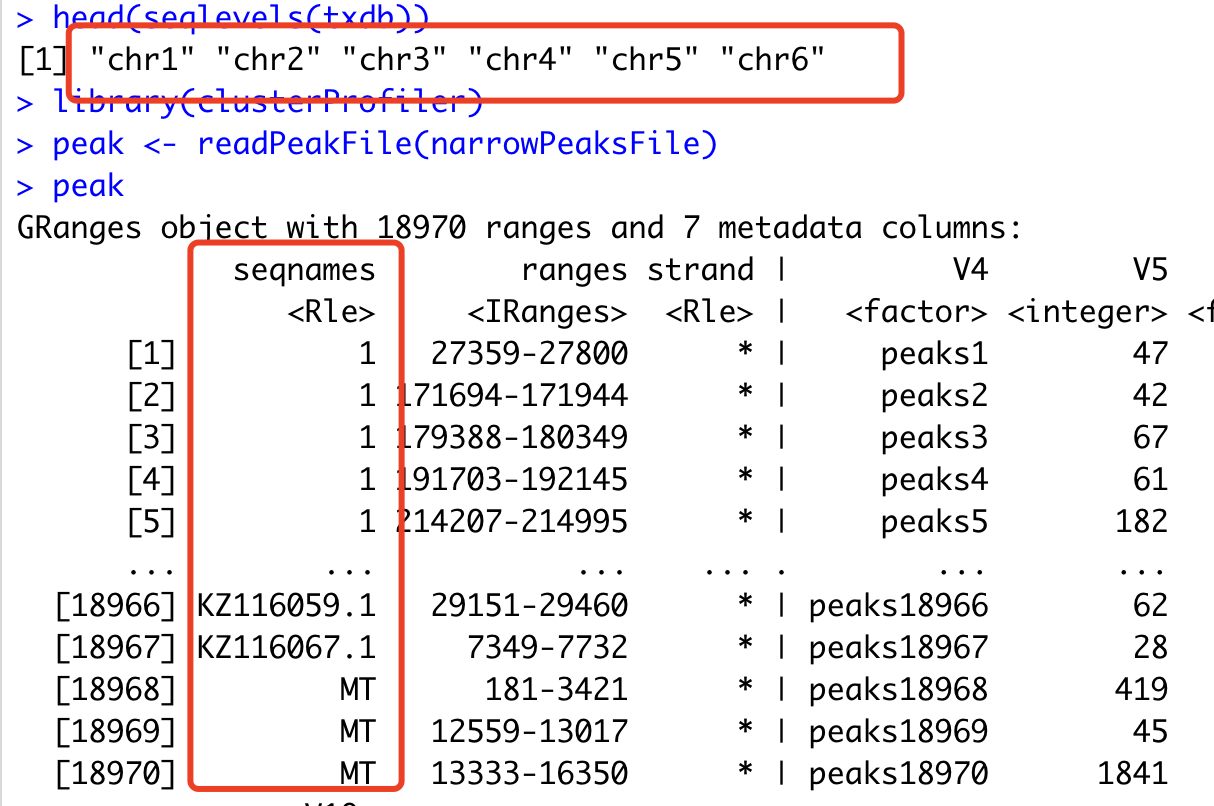
这里面的思考过程,就是我说的控制变量法,而且需要仔细检查每一个步骤的输入输出。
其实,这里面还有一个隐患,就是参考基因组版本的问题,因为我确实不知道学徒的上游流程,所以也没办法帮她检查,但是这些peaks除了可以注释到基因,还可以细致的看一下统计情况,如果参考基因组版本有问题,图表很容易看出来的。
peakAnno <- annotatePeak(peak, tssRegion=c(-3000, 3000),
TxDb=txdb, annoDb="org.Dr.eg.db")
peakAnno_df <- as.data.frame(peakAnno)
peakAnno_df[1:4,1:4]
promoter <- getPromoters(TxDb=txdb, upstream=3000, downstream=3000)
tagMatrix <- getTagMatrix(peak, windows=promoter)
# 然后查看这些peaks在所有基因的启动子附近的分布情况,热图模式
tagHeatmap(tagMatrix, xlim=c(-3000, 3000), color="red")
# 然后查看这些peaks在所有基因的启动子附近的分布情况,信号强度曲线图
plotAvgProf(tagMatrix, xlim=c(-3000, 3000),
xlab="Genomic Region (5'->3')", ylab = "Read Count Frequency")
plotAnnoPie(peakAnno)
比如,这个时候,拿到下面这样的图表,我就会初步认为是有问题的。
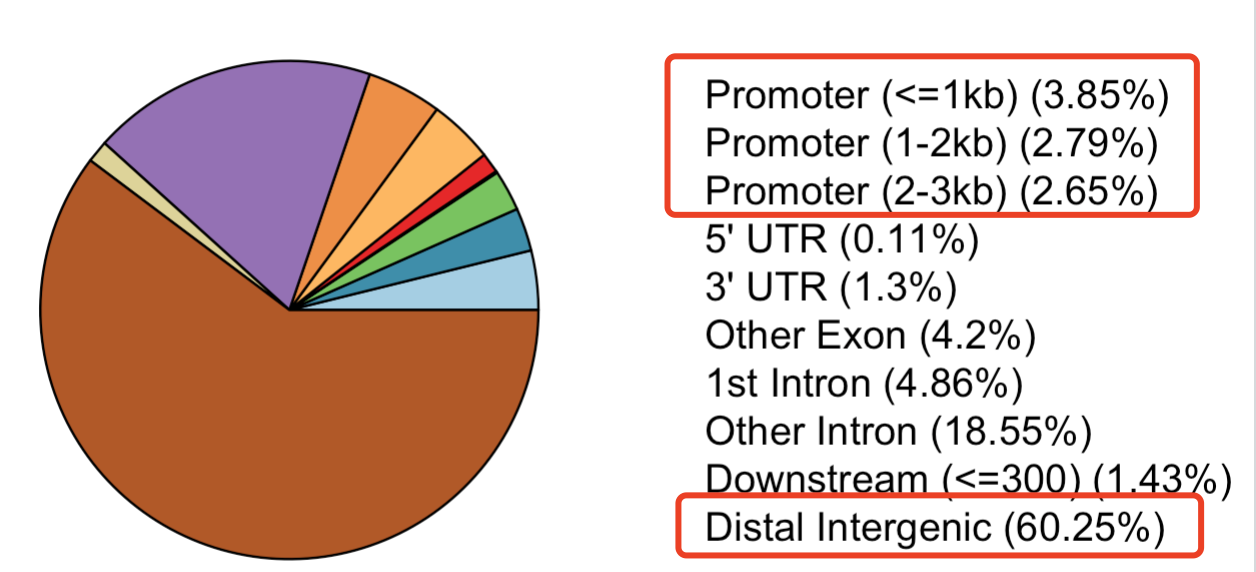
如果你想知道为什么,可能是需要看完我的全套表观教学视频了。