熟悉DNA测序的朋友应该是知道对肿瘤病人进行WES或者WGS等基因组测序后,可以得到成百上千的somatic突变,而这些突变都是ATCG碱基直接变化,所以它们变化的组合情况就6种,而加上上下文碱基也就96种,这96种碱基变化的比例的特殊组成,就是mutation signatures,而且COSMIC数据库上面有着30种已知的signatures,我们可以把这些signatures当做是一个有意义的生物学功能,这样一旦我们拿到自己的突变数据, 就可以通过非负矩阵分解的方法把自己的突变数据分解为这30个signatures的组合,那么如果我们拿到的是表达矩阵呢?
因为表达矩阵通常是bulk转录组测序,也就是说本来就是肿瘤细胞以及其肿瘤微环境的各种其他细胞组合而成,同理我们应该是可以根据表达量推断出来他们的细胞组分。当然,这个就需要算法上面的突破啦,下面我们就介绍一些相关方向的进展。
免疫微环境背景知识
低通量实验可以用免疫组化,免疫荧光和流式等方法来获得组织免疫细胞组成,而 bulk tumor转录组数据同样可以通过算法轻松得到肿瘤免疫细胞浸润水平,两篇综述系统性的整理和比较了主流算法。
- Hackl H, Charoentong P, Finotello F, et al. Computational genomics tools for dissecting tumour-immune cell interactions. Nat Rev Genet 2016;17:441-58.
- Finotello, Francesca, and Zlatko Trajanoski. Quantifying Tumor-Infiltrating Immune Cells from Transcriptomics Data. Cancer Immunology, Immunotherapy: CII 67, no. 7 (2018): 1031–40.
提到的算法主要是2大类,包括:
- 基于GSEA的半定量方法
- Deconvolution algorithms(分为partial deconvolution和complete deconvolution)
基于GSEA的半定量方法
基于GSEA半定量的方法最常用的就是ssGSEA(单样本GSEA),ssGSEA按照单个样本表达量的绝对值排序,计算特定gene set的累积经验概率分布,得到ES值代表这些基因在单个样本中协调上下调的程度,以此来代表免疫浸润细胞丰度。xCell,MCP-counter都是基于ssGSEA所开发出来的方法。
partial deconvolution
Deconvolution algorithms可以理解为一个基因在样本中的表达量是该基因在样本中不同细胞亚群表达水平和细胞分数权重的线性组合。近几年开发的CIBERSORT算法正是去卷积方法的应用。
TIMER量化6种免疫细胞,但是与CIBERSORT不同(CIBERSORT解析结果:22种免疫细胞相加的总占比为100%),TIMER没有把预测值标准化为1,故不可以把结果解释为细胞分数或是在不同的数据集中比较,还有EPIC,quanTIseq等等方法。
complete deconvolution
相比较与partial deconvolution,complete deconvolution不仅可以估计相对细胞分数同时还能disentangle表达谱。非负矩阵分解(NMF)就是一个很好的理解例子,但是NMF是完全无监督的方法,考虑到量化免疫细胞的要求,Gaujoux融合细胞marker genes的先验知识开发出半监督NMF,有兴趣了解的可以了解下CellMix这个R包。
量化工作中的challenges
Bulk tumor中,除了各类方法想要量化的细胞,还有一些未知的细胞。Deconvolution algorithms其中一个挑战就是对这些unknown tumor content的稳健性。EPIC和quanTIseq允许最终细胞分数总和低于1,来估计未知细胞分数。
此外,多重共线性也是deconvolution algorithm要考虑的问题,如基因表达在related cells中的高度相关。CIBERSORT,TIMER,quanTIseq采用了一系列的方法来减少共线性的影响。
今天,我们先一起来拆解一下CIBERSORT流程,配套R代码来自于 CIBERSORT R script v1.03 (last updated 07-10-2015),最新版是CIBERSORTX,我们下一次再解析。
相比较与基因芯片,RNA-seq数据具有“digital” nature和wider dynamic range使得量化工作更具有挑战性,目前只有EPIC和quanTIseq是针对RNAseq数据的。mRNA content,输入数据的统计学分布以及是否应该针对不同癌种,肿瘤类型(实体瘤和血液肿瘤)开发个性化的量化方法都是值得思考和改进的问题。
第一步,熟悉LM22和表达矩阵
代码如下,无需运行,我已经把两个矩阵保存为 ‘input.Rdata’ 文件啦
rm(list = ls())
options(stringsAsFactors = F)
# 首先读取两个文件
sig_matrix <-"LM22-ref.txt" # cibersoft 内置数据库挖掘
mixture_file <- "mRNA2.txt" # 约80M,TCGA数据库
# 两个表达矩阵需要取交集
#read in data
X <- read.table(sig_matrix,header=T,sep="\t",row.names=1,check.names=F)
Y <- read.table(mixture_file, header=T, sep="\t", check.names=F)
Y <- Y[!duplicated(Y[,1]),]
rownames(Y)<-Y[,1]
Y<-Y[,-1]
X <- data.matrix(X)
Y <- data.matrix(Y)
Y[1:4,1:4]
X[1:4,1:4]
dim(X)
dim(Y)
X <- X[order(rownames(X)),]
Y <- Y[order(rownames(Y)),]
#anti-log if max < 50 in mixture file
if(max(Y) < 50) {Y <- 2^Y}
QN = F
#quantile normalization of mixture file
if(QN == TRUE){
tmpc <- colnames(Y)
tmpr <- rownames(Y)
Y <- normalize.quantiles(Y)
colnames(Y) <- tmpc
rownames(Y) <- tmpr
}
#intersect genes
Xgns <- row.names(X)
Ygns <- row.names(Y)
YintX <- Ygns %in% Xgns
Y <- Y[YintX,]
XintY <- Xgns %in% row.names(Y)
X <- X[XintY,]
dim(X)
dim(Y)
#standardize sig matrix
X <- (X - mean(X)) / sd(as.vector(X))
Y[1:4,1:4]
X[1:4,1:4]
boxplot(X[,1:4])
save(X,Y,file = 'input.Rdata')
从代码里面,可以看到,LM22矩阵进行了zscore转换,待反卷积的表达矩阵虽然并没有被zscore,不过在后面的步骤仍然是会zscore的。
第二步,搞清楚 CoreAlg 函数
这里会使用被zscore后的LM22矩阵,来使用SVM算法来预测一个随机的Y变量。
rm(list = ls())
options(stringsAsFactors = F)
library(preprocessCore)
library(parallel)
library(e1071)
source("CIBERSORT.R")
load(file = 'input.Rdata')
Y[1:4,1:4]
X[1:4,1:4]
dim(X)
dim(Y)
# 下面的演示是为了搞清楚 CoreAlg 函数
# 并不需要保存任何信息
# 从表达矩阵Y里面,随机挑选LM22矩阵基因数量的表达量值
Ylist <- as.list(data.matrix(Y))
yr <- as.numeric(Ylist[ sample(length(Ylist),dim(X)[1]) ])
# yr 这个时候是一个假设的样本
#standardize mixture,就是scale 函数
yr <- (yr - mean(yr)) / sd(yr)
boxplot(yr)
# 每次随机挑选的yr,都是需要走后面的流程
# 一切都是默认值的支持向量机
# 这里的X是LM22矩阵,不同的免疫细胞比例组合成为不同的yr
# 这里的yr是随机的,反推免疫细胞比例
out=svm(X,yr)
out
out$SV
# SVM 需要自行学习哦
# 需要修改的参数包括:type="nu-regression",kernel="linear",nu=nus,scale=F
svn_itor <- 3
y=yr
res <- function(i){
if(i==1){nus <- 0.25}
if(i==2){nus <- 0.5}
if(i==3){nus <- 0.75}
model<-svm(X,y,type="nu-regression",kernel="linear",nu=nus,scale=F)
model
}
#Execute In a parallel way the SVM
if(Sys.info()['sysname'] == 'Windows') {
out <- mclapply(1:svn_itor, res, mc.cores=1)
}else {
out <- mclapply(1:svn_itor, res, mc.cores=svn_itor)
}
# 运行了Support Vector Machines,函数是 svm {e1071}
out
#Initiate two variables with 0
nusvm <- rep(0,svn_itor)
corrv <- rep(0,svn_itor)
t <- 1
while(t <= svn_itor) {
# 得到两个向量之间矩阵乘法的权重,此时应该只得到一个数字。
# 这样做是乘以系数
# 支持向量是数据集的点,它们靠近分隔类别的平面
# 现在的问题是,我没有任何类别(离散变量,例如“运动”、“电影”),但我有一个连续变量
mySupportVectors <- out[[t]]$SV
# 系数定义
myCoefficients <- out[[t]]$coefs
weights = t(myCoefficients) %*% mySupportVectors
# 设置权重和相关性
weights[which(weights<0)]<-0
w<-weights/sum(weights)
# 根据对应的权重与参考集相乘
u <- sweep(X,MARGIN=2,w,'*')
# 统计每行总和
k <- apply(u, 1, sum)
nusvm[t] <- sqrt((mean((k - y)^2)))
corrv[t] <- cor(k, y)
t <- t + 1
}
#pick best model
rmses <- nusvm
corrv
mn <- which.min(rmses)
mn
model <- out[[mn]]
# 从nus为0.25,0.5,0.75的3个模型里面挑选一个即可
#get and normalize coefficients
q <- t(model$coefs) %*% model$SV
q[which(q<0)]<-0
# w 就是计算后的22种免疫细胞的比例
w <- (q/sum(q))
mix_rmse <- rmses[mn]
mix_r <- corrv[mn]
# 会返回这个随机的y的免疫细胞组成情况,就是权重w
newList <- list("w" = w, "mix_rmse" = mix_rmse, "mix_r" = mix_r)
newList
# 根据对应的权重与参考集相乘
u <- sweep(X,MARGIN=2,w,'*')
k <- apply(u, 1, sum)
plot(y,k)
sqrt((mean((k - y)^2)))
cor(k, y)
# 通常这个预测结果都惨不忍睹
如果你硬是要去了解SMV原理,可能得去购买专业书籍了:

第3步,把CoreAlg函数运行1000次
每次随机挑选基因表达量,生成一个模拟的样本,然后使用zscore后的LM22矩阵经过SVM算法来预测:
rm(list = ls())
options(stringsAsFactors = F)
load(file = 'input.Rdata')
Y[1:4,1:4]
X[1:4,1:4]
dim(X)
dim(Y)
library(preprocessCore)
library(parallel)
library(e1071)
source("cibersort_ann.R")
itor <- 1
Ylist <- as.list(data.matrix(Y))
dist <- matrix()
# 就是把 CoreAlg 函数运行1000次
perm=1000
while(itor <= perm){
print(itor) # 打印进度
#random mixture
yr <- as.numeric(Ylist[ sample(length(Ylist),dim(X)[1]) ])
#standardize mixture
yr <- (yr - mean(yr)) / sd(yr)
#run CIBERSORT core algorithm
result <- CoreAlg(X, yr)
mix_r <- result$mix_r
#store correlation
if(itor == 1) {dist <- mix_r}
else {dist <- rbind(dist, mix_r)}
itor <- itor + 1
}
newList <- list("dist" = dist)
nulldist=sort(newList$dist)
# 这个nulldist 主要是用来计算P值
if(F){
P=perm
#empirical null distribution of correlation coefficients
if(P > 0) {nulldist <- sort(doPerm(P, X, Y)$dist)}
print(nulldist)
}
header <- c('Mixture',colnames(X),"P-value","Correlation","RMSE")
print(header)
save(nulldist,file = 'nulldist_perm_1000.Rdata')
load(file = 'nulldist_perm_1000.Rdata')
print(nulldist)
fivenum(print(nulldist))
每次预测的Y和随机生成的y差异都非常可怕,简直就是灾难现场!保存每次随机过程得到预测变量和随机变量的相关性系数,虽然都是0附近,因为预测的很糟糕,基本上都不相关。
> fivenum(nulldist)
[1] -0.079400079 -0.009724867 0.019402797 0.056885990 0.604810742
这个nulldist是1000次的相关性系数值,只有一个用处,就是计算后面真正的样本预测的P值而已。
第四步,对表达矩阵的每个样本进行SVM预测
理解了前面的代码,下面代码就非常容易理解,只不过是之前的预测变量y是随机的,这次是真实样本的基因表达量y值:
rm(list = ls())
options(stringsAsFactors = F)
load(file = 'input.Rdata')
Y[1:4,1:4]
X[1:4,1:4]
dim(X)
dim(Y)
library(preprocessCore)
library(parallel)
library(e1071)
source("cibersort_ann.R")
header <- c('Mixture',colnames(X),"P-value","Correlation","RMSE")
print(header)
load(file = 'nulldist_perm_1000.Rdata')
output <- matrix()
itor <- 1
mix <- dim(Y)[2]
pval <- 9999
P=1000
# 表达矩阵的每个样本,都需要计算一下LM22的比例
#iterate through mix
while(itor <= mix){
##################################
## Analyze the first mixed sample
##################################
y <- Y[,itor]
#标准化样本数据集
y <- (y - mean(y)) / sd(y)
#执行SVR核心算法
result <- CoreAlg(X, y)
#获得结果
w <- result$w
mix_r <- result$mix_r
mix_rmse <- result$mix_rmse
#计算p-value
if(P > 0) {pval <- 1 - (which.min(abs(nulldist - mix_r)) / length(nulldist))}
#输出output
out <- c(colnames(Y)[itor],w,pval,mix_r,mix_rmse)
if(itor == 1) {output <- out}
else {output <- rbind(output, out)}
itor <- itor + 1
}
head(output)
#save results
write.table(rbind(header,output), file="CIBERSORT-Results.txt", sep="\t", row.names=F, col.names=F, quote=F)
#return matrix object containing all results
obj <- rbind(header,output)
obj <- obj[,-1]
obj <- obj[-1,]
obj <- matrix(as.numeric(unlist(obj)),nrow=nrow(obj))
rownames(obj) <- colnames(Y)
colnames(obj) <- c(colnames(X),"P-value","Correlation","RMSE")
obj
save(obj,file = 'output_obj.Rdata')
第五步,可视化cibersoft去卷积结果
结果很容易理解,就是每个样本的各个免疫细胞比例而已,再加上 “P.value”,”Correlation”,”RMSE” 这3列:
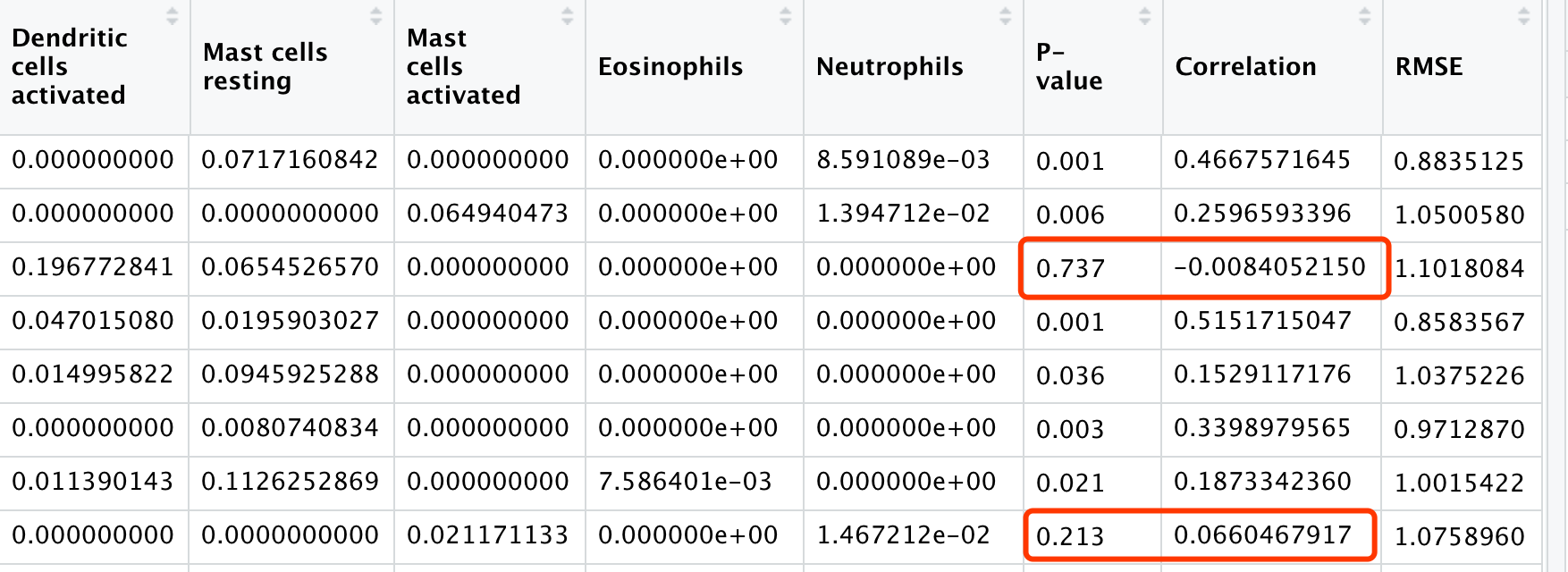
每个样本的各个免疫细胞比例矩阵就可以进行一些可视化,主要是参考CIBERSORT官网绘制:
可视化1:热图展现预测的22种免疫细胞占比
这个例子的408个样本的22种免疫细胞比例热图展现如下:
因为22种免疫细胞有一些在过半的样本里面含量为0,就剔除了,无需可视化展现
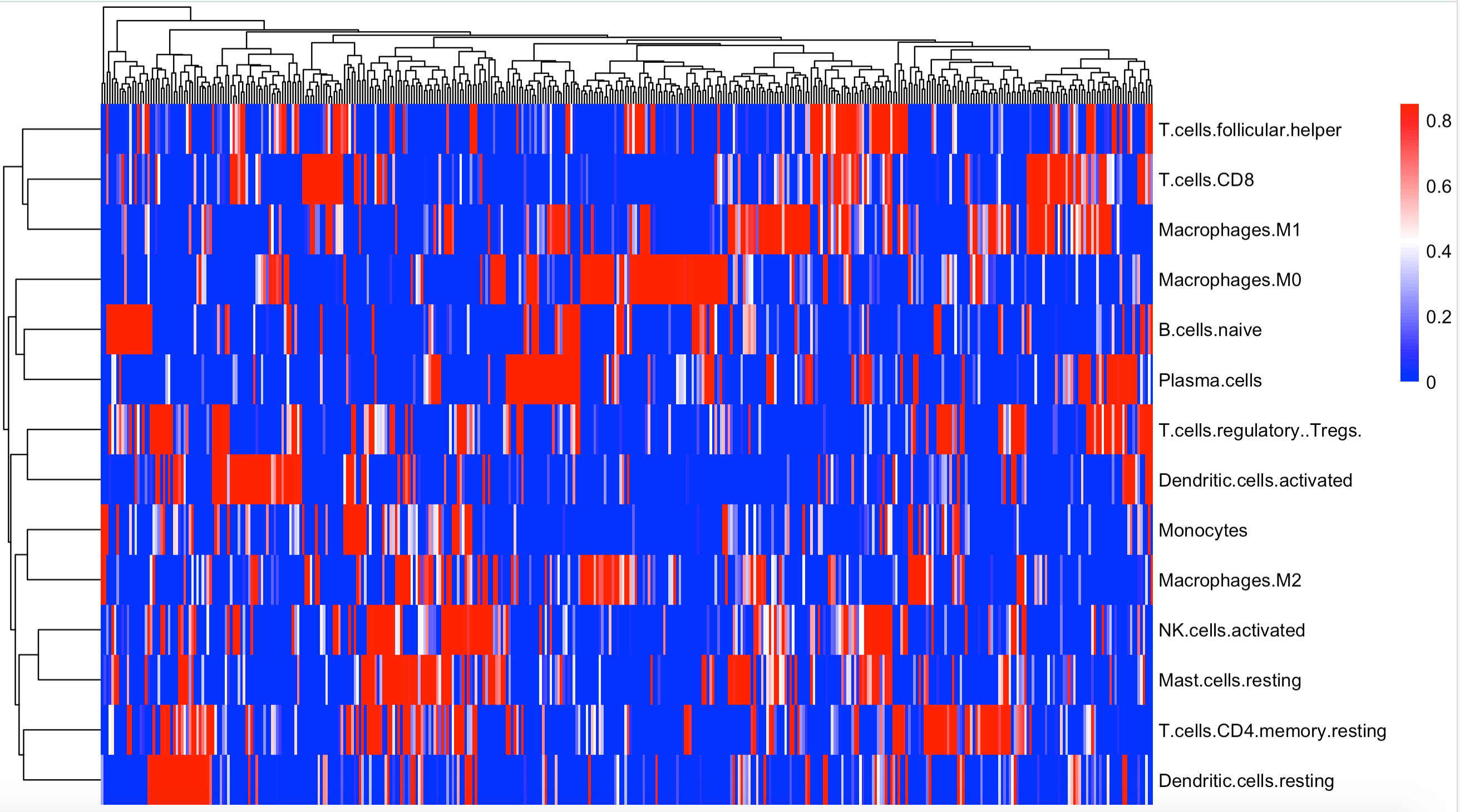
可视化2:柱状图展现预测的22种免疫细胞占比
这个时候不过滤了:
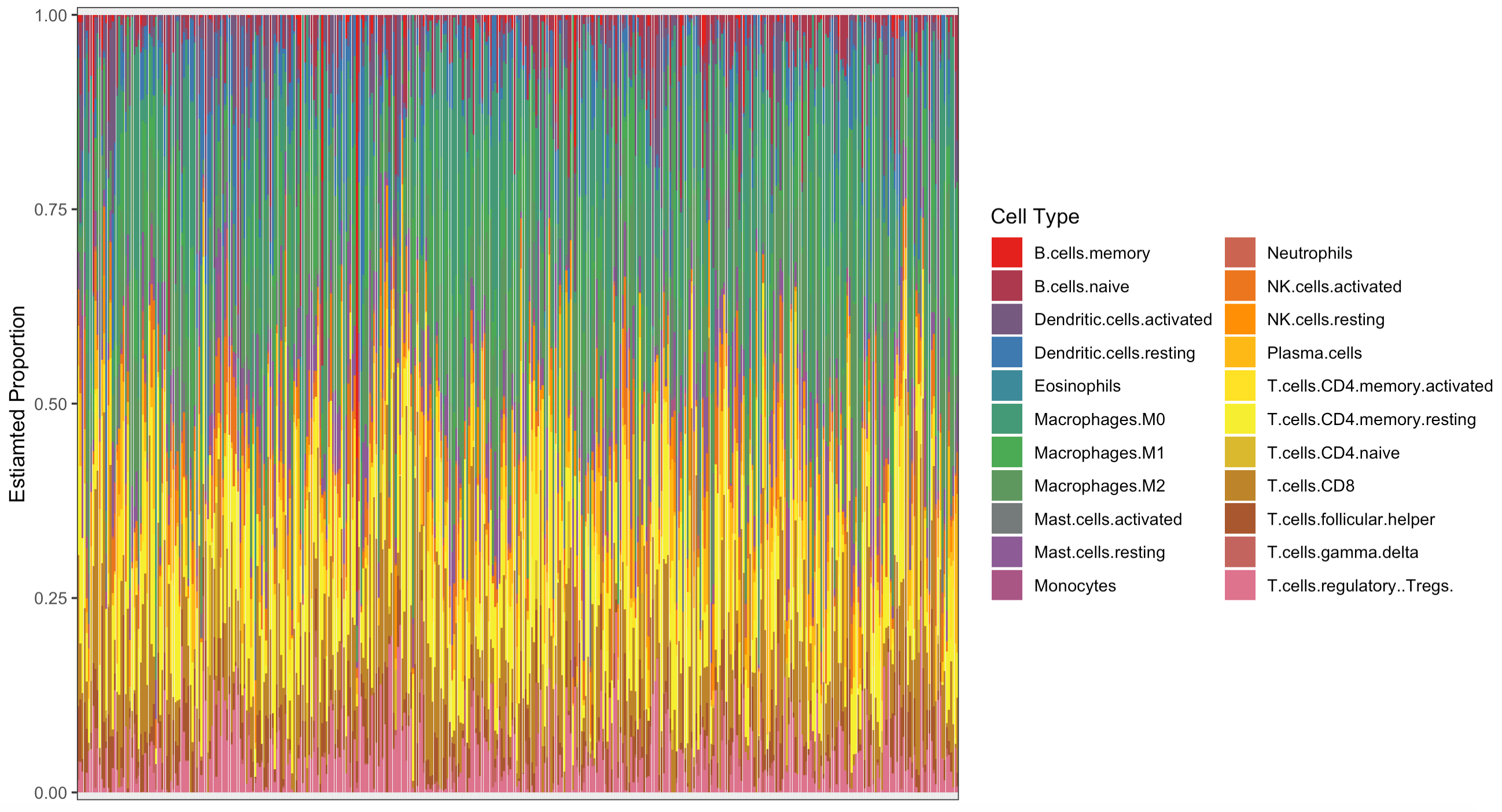
可视化3:箱线图展现预测的22种免疫细胞占比
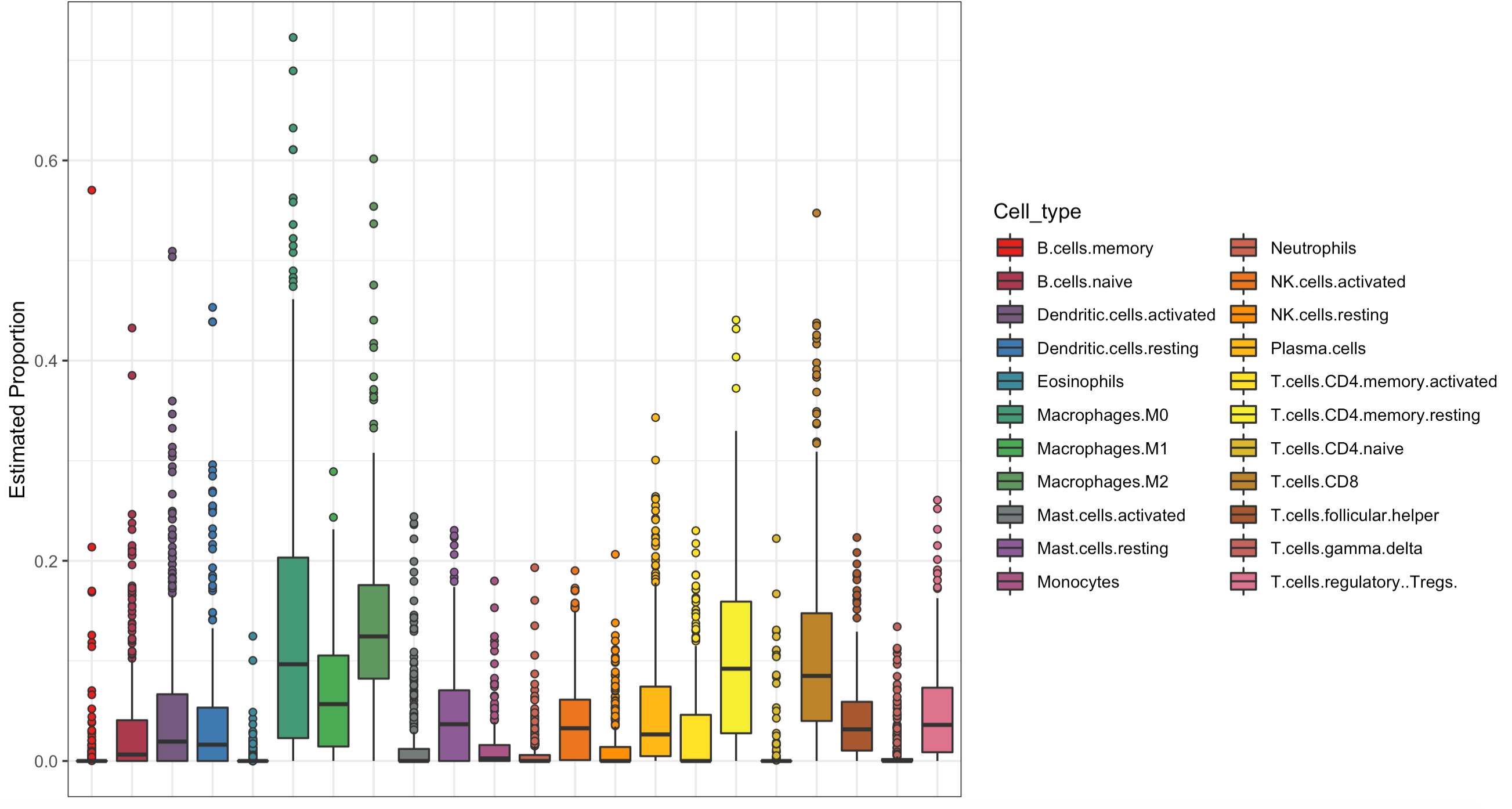
这个CIBERSORT来反推免疫细胞浸润情况可能是比较难理解,但是我们以前也分享过 纯R代码实现ssGSEA算法评估肿瘤免疫浸润程度,就很容易理解了,仅仅是ssGSEA得分。
文末友情宣传
强烈建议你推荐我们生信技能树给身边的博士后以及年轻生物学PI,帮助他们多一点数据认知,让科研更上一个台阶:
- 生信爆款入门-全球听(买一得五)(第4期),你的生物信息学入门课
- 数据挖掘第2期(两天变三周,实力加量),医学生/临床医师首选技能提高课
- 生信技能树的2019年终总结 ,你的生物信息学成长宝藏
- 2020学习主旋律,B站74小时免费教学视频为你领路,还等什么,看啊!!!
付费内容分割线
为有效杜绝黑粉跟我扯皮,设置一个付费分割线,这样它们就没办法复制粘贴我的代码,也不可能给我留言骂街了!(付费6元,拿到CIBERSORT流程的测试数据和代码)
世界顿时清净很多!
综合脚本
链接:https://share.weiyun.com/5HaTRjs 密码:
这个微云链接需要密码,付费6元可以得到,请移步到微信公众号 https://mp.weixin.qq.com/s/rK7FFQuEPKpEU6qYbVB4Rw 付费获取哈!
运行CIBERSORT.R脚本
其实就一句代码:CIBERSORT(sig_matrix, mixture_file, perm=10, QN=TRUE)
但是需要自己定义和整理好表达矩阵txt文件,会R语言的都很容易看懂的。
rm(list = ls())
options(stringsAsFactors = F)
load(file = 'input.Rdata')
Y[1:4,1:4]
X[1:4,1:4]
dim(X)
dim(Y)
library(preprocessCore)
library(parallel)
library(e1071)
source("CIBERSORT.R")
# 运行CIBERSORT官方函数,获得三个官方算法定义的Function:
# CIBERSORT,CoreAlg和doPerm。
sig_matrix = 'LM22-ref.txt'
mixture_file = 'test.txt'
# 主要是 read.table 的参数可以随意修改,根据你自己的表达矩阵txt文件适应性调整即可
X <- read.table(sig_matrix,header=T,sep="\t",row.names=1,check.names=F)
Y <- read.table(mixture_file, header=T, sep="\t", check.names=F)
# 运行 CIBERSORT 会写出 CIBERSORT-Results.txt 文件覆盖掉之前的结果
# 务必看清楚 test.txt 这个表达矩阵格式哦!!!
# CIBERSORT(sig_matrix, mixture_file, perm=10, QN=TRUE)
# 后面可以对 CIBERSORT-Results.txt 里面的结果进行各式各样的可视化