前面我们一起学习了单细胞转录组数据的上游分析,而且了解了自己的项目的样本数量和测序量,还过滤了不合格的细胞和基因, 系列教程目录如下:
- 01. 上游分析流程
- 02. 课题多少个样品,测序数据量如何
- 03. 过滤不合格细胞和基因(数据质控很重要)
- 04. 过滤线粒体核糖体基因
- 05. 去除细胞效应和基因效应
理论上我们已经足够认识表达矩阵了,现在可以开始单细胞转录组数据的主角:降维聚类分群。如果你的背景知识不足,也可以先读一下综述,我们单细胞天地有中文指引: - 单细胞RNA-seq数据分析最佳实践(上)
- 单细胞RNA-seq数据分析最佳实践(中)
- 单细胞RNA-seq数据分析最佳实践(下)
降维聚类分群是一条龙分析
我们并不是开发单细胞数据处理算法,所以大概率上,大家其实会把降维聚类分群一起做了,在seurat3里面的代码是:
sce <- NormalizeData(sce, normalization.method = "LogNormalize", scale.factor = 10000) sce <- FindVariableFeatures(sce, selection.method = "vst", nfeatures = 2000) # 步骤 ScaleData 的耗时取决于电脑系统配置(保守估计大于一分钟) sce <- ScaleData(sce) sce <- RunPCA(object = sce, pc.genes = VariableFeatures(sce)) sce <- FindNeighbors(sce, dims = 1:15) sce <- FindClusters(sce, resolution = 0.2) # 这个 resolution 可以调整,值越大,分出来的细胞亚群越多,默认是 0.8 table(sce@meta.data$RNA_snn_res.0.2)首先看两种降维
简单解释一下,这代码里面的FindVariableFeatures和RunPCA函数,是两种不同策略的降维。
- 首先FindVariableFeatures是硬过滤,根据一些统计指标,比如sd,mad,vst等等来判断你输入的单细胞表达矩阵里面的2万多个基因里面,最重要的2000个基因,其余的1.8万个基因下游分析就不考虑了。
- 然后RunPCA函数,其实跑完之后2000个基因会转变为2000个维度,但是我们通常看前面的十几个维度就ok了,所以也是一个效率非常高的降维方式。
这个时候,你一定有疑问,为什么FindVariableFeatures是挑选2000个基因而不是其它数量呢?RunPCA函数跑完后我们应该是挑选前多少个维度呢?10个还是15个,还是20个还是50呢。然后看聚类分群
聚类分群是紧密连接的,细胞可以看做是空间的不同点,如果是二维平面空间,点与点之间的距离很方便计算,距离的远近就决定着细胞是否属于一个类群。
计算距离的公式很多,我们就不一一展开,但是需要注意的是二维平面空间,三维球体空间的细胞距离很方便计算,但是如果是50个维度的空间,计算几万个细胞之间的距离就很可怕了,如果是2000个维度,甚至是2万个维度,基本上个人计算机就可以放弃了。这就是为什么我们前面通常是需要降维的。
下面是一个典型的单细胞转录组项目数据处理的描述:

可以看到他们的第一步降维是,选取top 5000的表达量离散度大的基因,第二步降维是选取top20的主成分。使用KNN-graph的聚类,最终定下来了10个细胞亚群。
一般来说,如果单细胞转录组数据仅仅是文章生物学故事的一个环节,就会采取标准的seurat流程,如下所示:

如果你看的文献足够多,还会发现,在降维聚类分群之后,通常是有一个细胞在二维平面的散点图展示,如下所示:
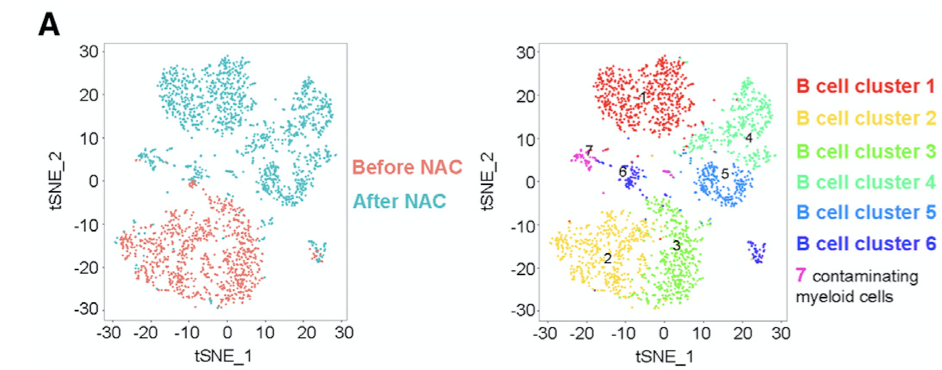
如果你足够心细,也会发现其实细胞的空间距离排布坐标通常是tSNE和umap来展现。那我们说的tSNE和umap是怎么回事呢
tSNE(t-distributed stochastic neighbor embedding) 是一种非线性降维的算法,是流形学习的一种,当然,你大概率上是无需理解流形学习的,认识一下其它流形学习的经典方法即可,包括:Isomap,LLE,LE和diffusion maps等。我发现这篇推文介绍的非常好:单细胞中的流形(一): 理解 tSNE中的perplexity,看完你需要记住的是;
- 困惑度(perplexity)可以表示细胞的邻近个数,在tSNE图上的直观反映是细胞点的分布是否紧凑。perplexity设置越大,细胞分布越紧凑。
- tSNE的参数设置:perplexity < (细胞数-1)/3,建议perplexity = 细胞数 / 50;
- tSNE倾向于保留数据的局部结构。
本来我还期待着他们继续讲解umap,但是看起来似乎公众号已经停更了。唉,现在搞分享,写原创教程也挺不容易的, 很容易就quit了。
我给大家的策略是,反正你得多尝试,umap啥都尝试一遍,最后选择效果好的图表去展现即可。可以修改的参数
第一次降维,表达矩阵里面通常是2~3万的基因数量,需要筛选到千这个数量级,可以是1000-5000,都是可以的。
文末友情宣传
强烈建议你推荐给身边的博士后以及年轻生物学PI,多一点数据认知,让他们的科研上一个台阶:
- 全国巡讲全球听(买一得五)第3期(4月6日开始) ,你的生物信息学入门课
- 生信技能树的2019年终总结 ,你的生物信息学成长宝藏
- 2020学习主旋律,B站74小时免费教学视频为你领路