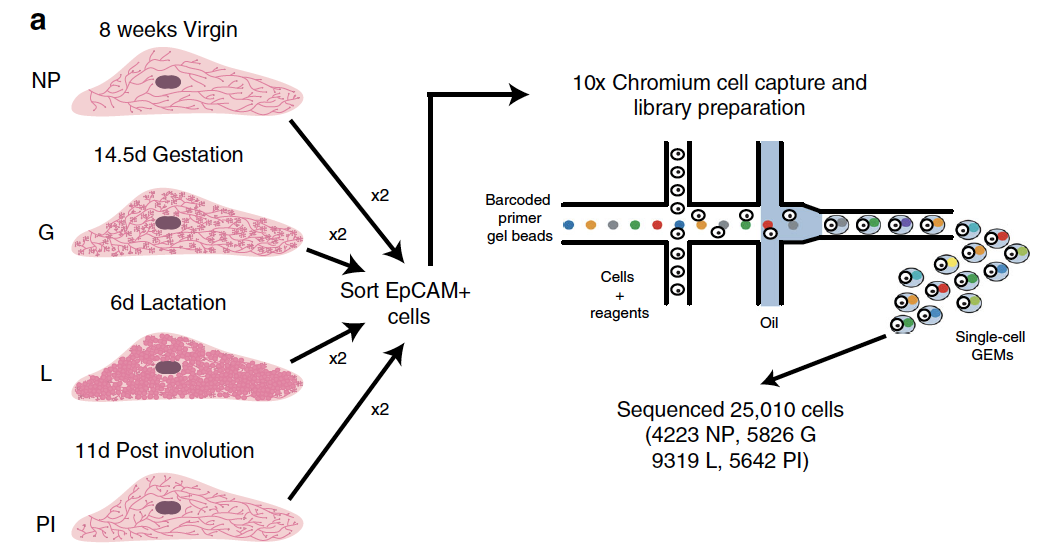
发表在2017年10月的NC文章:Differentiation dynamics of mammary epithelial cells revealed by single-cell RNA sequencing 用10X单细胞转录组测序来探索小鼠的乳腺发育情况,包括了4个发育阶段:
- nulliparous (NP) 未怀孕时期
- day 14.5 gestation (G) 妊娠期第14.5天
- day 6 lactation (L) 哺乳期第6天
- 11 days post natural involution (PI) 完全自然退化期第11天
每个发育时期选取两个样本,全部的是8个10X单细胞转录组样本,如下所示:
全部数据在:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE106273 可以下载!
8个10X单细胞转录组样本的数据分别是:
GSM2834498 NP_1
GSM2834499 NP_2
GSM2834500 G_1
GSM2834501 G_2
GSM2834502 L_1
GSM2834503 L_2
GSM2834504 PI_1
GSM2834505 PI_2
作者给出了全部样本的表达矩阵:
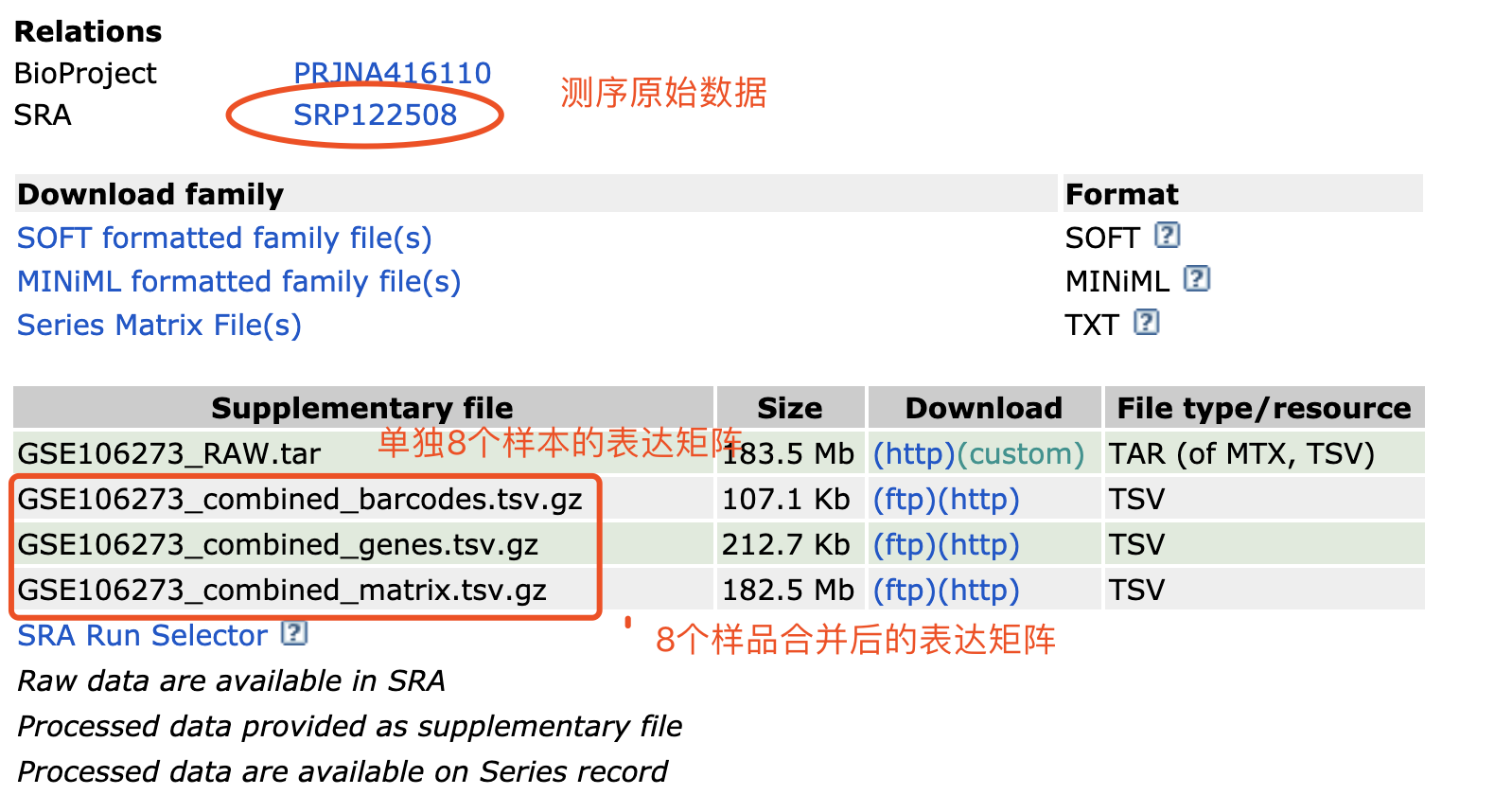
文章里面写的很清楚,使用的是 cellranger count 对10X单细胞转录组数据进行定量,然后使用的是 cellranger aggr 进行合并多个样本。
The reads were aligned to the mm10 reference genome using a pre-built annotation package obtained from the 10X Genomics website.
All lanes per sample were processed using the ‘cellranger count’ function.
The output from different lanes was then aggregated using ‘cellranger aggr’ with –normalise set to ‘none’.
考虑到,我们这里如果重新下载10X测序数据,走cellranger流程是一个力气活,这里我们就演示如何使用seurat包来进行多样本合并吧!如果你确实感兴趣cellranger流程,我们在单细胞天地多次分享过流程笔记,大家可以自行前往学习,如下:
- 单细胞实战(一)数据下载
- 单细胞实战(二) cell ranger使用前注意事项
- 单细胞实战(三) Cell Ranger使用初探
- 单细胞实战(四) Cell Ranger流程概览
- 单细胞实战(五) 理解cellranger count的结果
因为这里我们重点介绍seurat包来进行多样本合并,所以大家最好是有基础知识,比如听完我的全网第一个单细胞课程(基础)满一千份销量就停止发售 内容,就是一些R包的认知,包括 scater,monocle,Seurat,scran,M3Drop 需要熟练掌握它们的对象,:一些单细胞转录组R包的对象 ,分析流程也大同小异:
- step1: 创建对象
- step2: 质量控制
- step3: 表达量的标准化和归一化
- step4: 去除干扰因素(多个样本整合)
- step5: 判断重要的基因
- step6: 多种降维算法
- step7: 可视化降维结果
- step8: 多种聚类算法
- step9: 聚类后找每个细胞亚群的标志基因
- step10: 继续分类
在seurat3里面多个10X单细胞转录组整合其实就一个merge函数而已
首先分开读取全部的8个10X单细胞转录组数据
需要下载的文件有点大,约200M,所以如果在中国大陆,通常是会下载失败,大家加油哦。链接是:https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSE106273&format=file
成功下载并且解压后如下:
9.9K Oct 27 2017 GSM2834498_NP_1_barcodes.tsv.gz
213K Oct 27 2017 GSM2834498_NP_1_genes.tsv.gz
21M Oct 27 2017 GSM2834498_NP_1_matrix.mtx.gz
9.5K Oct 27 2017 GSM2834499_NP_2_barcodes.tsv.gz
213K Oct 27 2017 GSM2834499_NP_2_genes.tsv.gz
18M Oct 27 2017 GSM2834499_NP_2_matrix.mtx.gz
13K Oct 27 2017 GSM2834500_G_1_barcodes.tsv.gz
213K Oct 27 2017 GSM2834500_G_1_genes.tsv.gz
26M Oct 27 2017 GSM2834500_G_1_matrix.mtx.gz
13K Oct 27 2017 GSM2834501_G_2_barcodes.tsv.gz
213K Oct 27 2017 GSM2834501_G_2_genes.tsv.gz
26M Oct 27 2017 GSM2834501_G_2_matrix.mtx.gz
24K Oct 27 2017 GSM2834502_L_1_barcodes.tsv.gz
213K Oct 27 2017 GSM2834502_L_1_genes.tsv.gz
30M Oct 27 2017 GSM2834502_L_1_matrix.mtx.gz
15K Oct 27 2017 GSM2834503_L_2_barcodes.tsv.gz
213K Oct 27 2017 GSM2834503_L_2_genes.tsv.gz
21M Oct 27 2017 GSM2834503_L_2_matrix.mtx.gz
6.9K Oct 27 2017 GSM2834504_PI_1_barcodes.tsv.gz
213K Oct 27 2017 GSM2834504_PI_1_genes.tsv.gz
11M Oct 27 2017 GSM2834504_PI_1_matrix.mtx.gz
18K Oct 27 2017 GSM2834505_PI_2_barcodes.tsv.gz
213K Oct 27 2017 GSM2834505_PI_2_genes.tsv.gz
29M Oct 27 2017 GSM2834505_PI_2_matrix.mtx.gz
如果你查看 Read10X 函数 ,会发现它只能是接受一个文件夹,然后在文件夹里面选择下面的4个文件名:
barcode.loc <- file.path(run, "barcodes.tsv")
gene.loc <- file.path(run, "genes.tsv")
features.loc <- file.path(run, "features.tsv.gz")
matrix.loc <- file.path(run, "matrix.mtx")
所以我们需要把上面8个样本的24个文件,存放到8个文件夹里面去,被 Read10X 函数读取。这个时候的预处理代码,跟单细胞无关,而且比较复杂,如果你无法理解也不要勉强,总之就是把文件夹层级结构做好即可:
library(Seurat)
fs=list.files('./','^GSM')
library(stringr)
samples=str_split(fs,'_',simplify = T)[,1]
lapply(unique(samples),function(x){
y=fs[grepl(x,fs)]
folder=paste(str_split(y[1],'_',simplify = T)[,2:3],collapse = '')
dir.create(folder,recursive = T)
#file.rename(y[1],file.path(folder,"barcodes.tsv.gz"))
file.rename(y[2],file.path(folder,"genes.tsv.gz"))
file.rename(y[3],file.path(folder,"matrix.mtx.gz"))
})
# > folders=list.files('./',pattern='[12]$')
# > folders
# [1] "G1" "G2" "L1" "L2" "NP1" "NP2" "PI1" "PI2"
# >
文件夹整理后大概是下面这样的结构:
├── 10x.Rproj
├── G1
│ ├── barcodes.tsv.gz
│ ├── genes.tsv.gz
│ └── matrix.mtx.gz
├── G2
│ ├── barcodes.tsv.gz
│ ├── genes.tsv.gz
│ └── matrix.mtx.gz
├── L1
│ ├── barcodes.tsv.gz
│ ├── genes.tsv.gz
│ └── matrix.mtx.gz
├── L2
│ ├── barcodes.tsv.gz
│ ├── genes.tsv.gz
│ └── matrix.mtx.gz
├── NP1
│ ├── barcodes.tsv.gz
│ ├── genes.tsv.gz
│ └── matrix.mtx.gz
├── NP2
│ ├── barcodes.tsv.gz
│ ├── genes.tsv.gz
│ └── matrix.mtx.gz
├── PI1
│ ├── barcodes.tsv.gz
│ ├── genes.tsv.gz
│ └── matrix.mtx.gz
├── PI2
│ ├── barcodes.tsv.gz
│ ├── genes.tsv.gz
│ └── matrix.mtx.gz
└── run.R
值得注意的是,本来我以为,是可以读取gz压缩格式的文件,后来发现不是我想的那样,还是需要解压,所以我写了一个shell命令做这件事,在mac电脑可以使用,如果你是Windows,建议安装git软件后,在git bash里面运行这个命令:
ls */*gz|xargs gunzip
批量读取8个10X单细胞转录组数据文件夹:
folders=list.files('./',pattern='[12]$')
folders
library(Seurat)
sceList = lapply(folders,function(folder){
CreateSeuratObject(counts = Read10X(folder),
project = folder )
})
然后运行merge函数
sce.big <- merge(sceList[[1]],
y = c(sceList[[2]],sceList[[3]],sceList[[4]],sceList[[5]],
sceList[[6]],sceList[[7]],sceList[[8]]),
add.cell.ids = folders,
project = "mouse8")
sce.big
# > sce.big
# An object of class Seurat
# 27998 features across 25806 samples within 1 assay
# Active assay: RNA (27998 features)
# >
table(sce.big$orig.ident)
save(sce.big,file = 'sce.big.merge.mouse8.Rdata')
最后查看merge前后分群的结果对比
首先需要降维聚类分群,代码比较简单,如下:
load(file = 'sce.big.merge.mouse8.Rdata')
library(Seurat)
# 步骤 ScaleData 的耗时取决于电脑系统配置(保守估计大于一分钟)
sce.big <- ScaleData(object = sce.big,
vars.to.regress = c('nCount_RNA'),
model.use = 'linear',
use.umi = FALSE)
sce.big <- FindVariableFeatures(object = sce.big,
mean.function = ExpMean,
dispersion.function = LogVMR,
x.low.cutoff = 0.0125,
x.high.cutoff = 4,
y.cutoff = 0.5)
length(VariableFeatures(sce.big))
sce.big <- RunPCA(object = sce.big, pc.genes = VariableFeatures(sce.big))
# 下面只是展现不同降维算法而已,并不要求都使用一次。
sce.big <- RunICA(sce.big )
sce.big <- RunTSNE(sce.big )
#sce.big <- RunUMAP(sce.big,dims = 1:10)
#VizPCA( sce.big, pcs.use = 1:2)
DimPlot(object = sce.big, reduction = "pca")
DimPlot(object = sce.big, reduction = "ica")
DimPlot(object = sce.big, reduction = "tsne")
tSNE降维后可视化,感觉图看起来还蛮酷炫
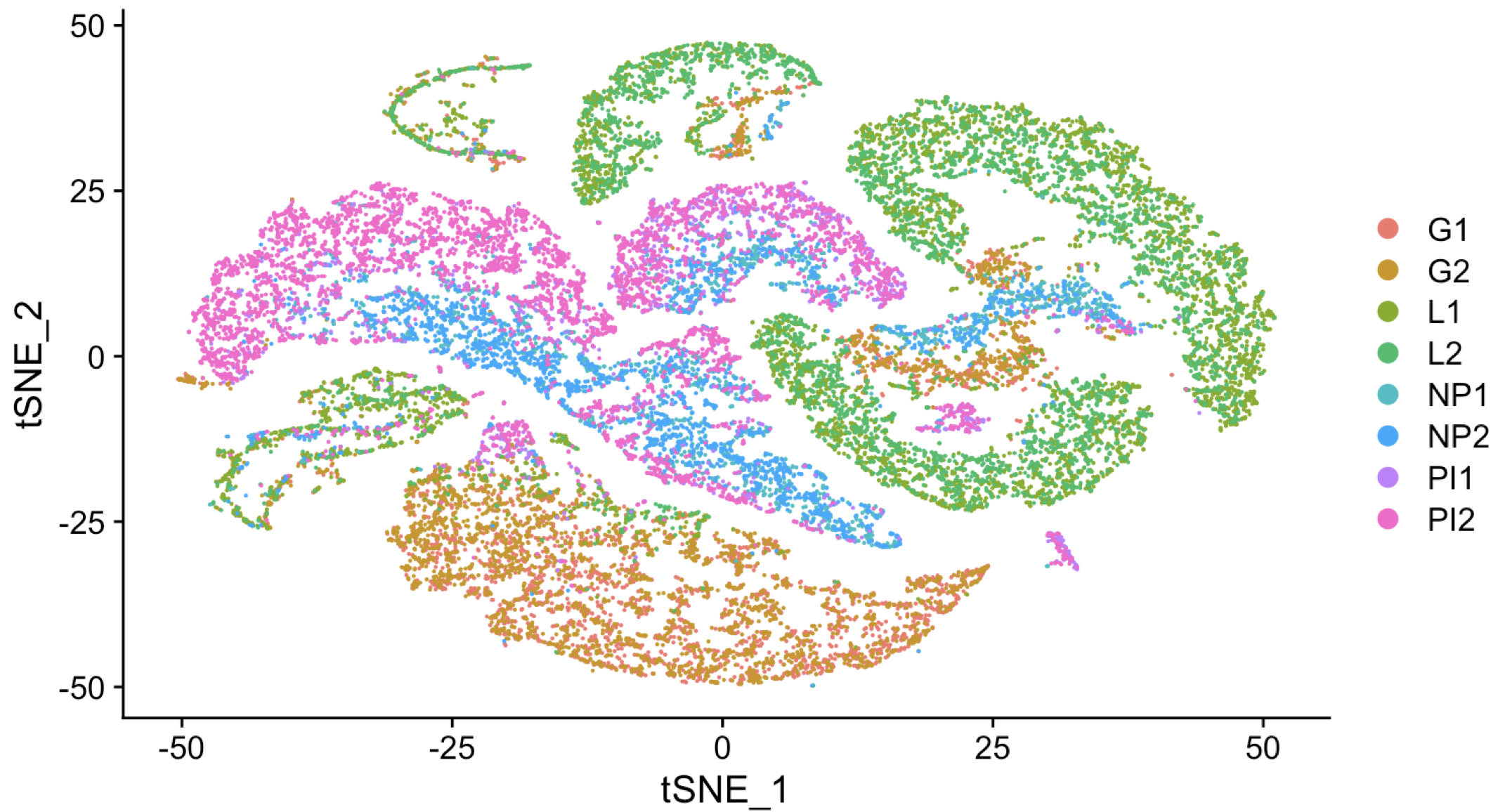
分群的话,我这里尊重原文,也采纳15个群,所以调整了 resolution 参数:
# 针对PCA降维后的表达矩阵进行聚类 FindNeighbors+FindClusters 两个步骤。
sce.big <- FindNeighbors(object = sce.big, dims = 1:20, verbose = FALSE)
sce.big <- FindClusters(object = sce.big, resolution = 0.3,verbose = FALSE)
# 继续tSNE可视化
DimPlot(object = sce.big, reduction = "tsne",group.by = 'RNA_snn_res.0.3')
DimPlot(object = sce.big, reduction = "tsne",
group.by = 'orig.ident')
table(sce.big$orig.ident,sce.big@meta.data$RNA_snn_res.0.3)
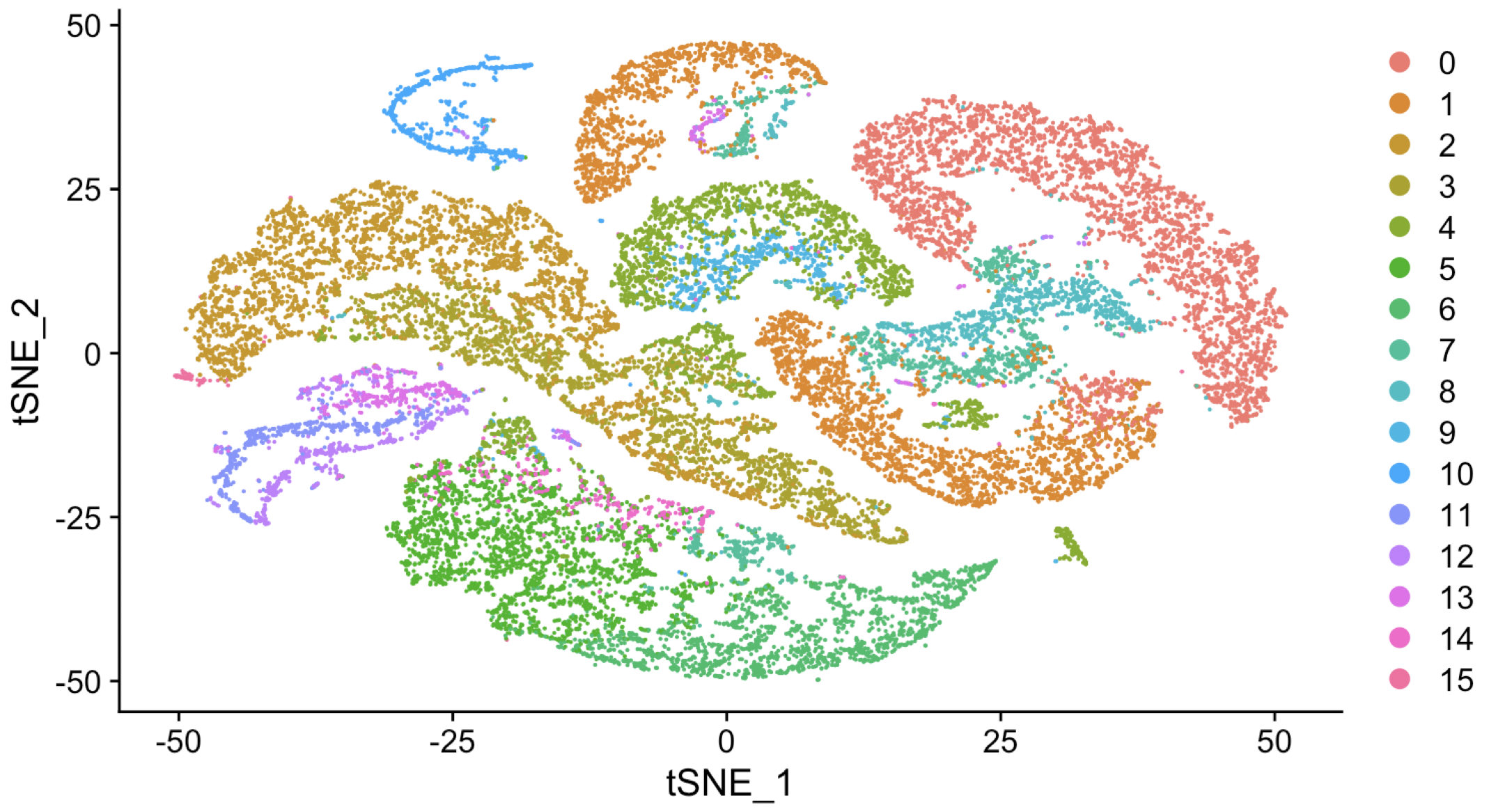
可以看到,不同的细胞亚群,其实就分散在不同的样本里面,当然了,同一个发育时期的两个样本倒是拆的很开,所以可以看出来我们的单细胞转录组样本批次效应去除的很好,只不过呢,不同发育时期,我们的细胞差异真的是太大了,所以它们被划分成为了截然不同的群体。

是不是很有趣,是不是感觉自己对批次效应,和细胞真实差异的理解更进了一步呢?
原文
通过调整及采纳合适的参数,原文把细胞区分成为15个细胞亚群,如下所示:
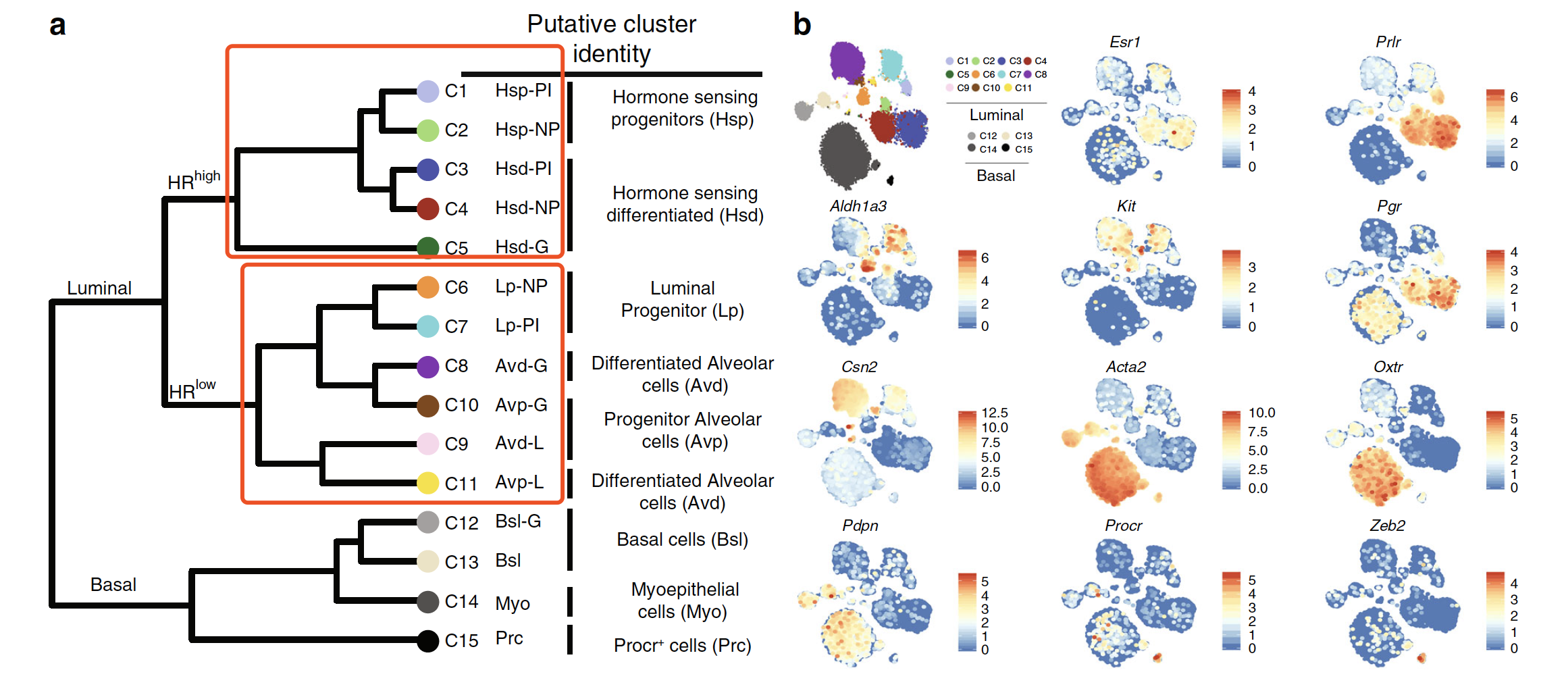
可以看到我们的结果是差不多的,从细胞亚群的数量上来看,L发育时期是2个主群,PI发育时期是2个主群,G发育时期也是3个主群,NP也是3个主群,然后这4个发育时期,都是有一些细胞分给了basal,是4个主亚群。
文末友情宣传
强烈建议你推荐给身边的博士后以及年轻生物学PI,多一点数据认知,让他们的科研上一个台阶:
- 全国巡讲全球听(买一得五) ,你的生物信息学入门课
- 生信技能树的2019年终总结 ,你的生物信息学成长宝藏
- 2020学习主旋律,B站74小时免费教学视频为你领路