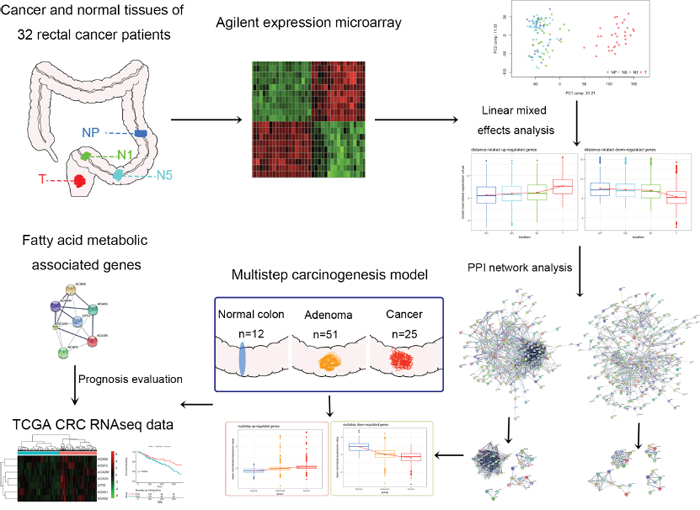
本期我们学习的文章是Integrated transcriptomic analysis of distance-related field cancerization in rectal cancer patients ,文章思路如下图所示:取肿瘤不同距离处样本,测序,找到表达量与距肿瘤远近相关的基因,然后进行功能注释、生存分析等等。
学习任务是:用lme4构建线性混合效应模型,找到表达量与距离相关的基因。
为什么要用线性混合效应模型
我们希望找到表达量与距离相关的基因,但因为来自同一个患者的表达量是相关的,所以这128个样本不满足普通线性回归的独立性假设,需要用到线性混合效应模型。
下载数据
step1.从GEO下载表达数据和表型信息
rm(list = ls())
### 下载GEO数据
library(GEOquery)
gset <- getGEO('GSE90627', destdir="./raw-data/",
AnnotGPL = F, ## 注释文件
getGPL = F)
gset
#注释文件
gpl <- getGEO("GPL17077",destdir="./raw-data/")
ids <- Table(gpl)
### 取表达矩阵
a=gset[[1]]
expr=exprs(a)
dim(expr)
expr[1:4,1:4]
#把探针号换成gene symbol
rownames(expr) <- ids[match(rownames(expr),ids$ID),2]
### 表型信息
pd=pData(a)
pd <- pd[,c(1,2,37:40)]
colnames(pd)[3:6] <- c("age","gender","patient","tissue")
# 构建出patient和distance变量
library(stringr)
pd$distance <- str_split(pd$title,"_",simplify = T)[,2]
pd$distance[97:128] <- rep("0cm",32)
pd$patient <- str_split(pd$title,"_",simplify = T)[,3]
pd$patient[97:128] <- str_split(pd$title[97:128],"_",simplify = T)[,2]
str(pd) #查看数据结构
save(expr,pd,file = "./Rdata/GSE90627_eSet.Rdata")
step2.文章的Supplementary Figure 1记录了每个患者的末端样本距离,复制到excel中,保存为sTable1.csv
rm(list=ls())
load("./Rdata/GSE90627_eSet.Rdata")
#读入取样末端距离数据
NP <- read.csv("./raw-data/sTable1.csv",header = T, stringsAsFactors = F)
#将距离改为数字
pd$distance2 <- substr(pd$distance,1,1)
pd$distance2[1:32] <- NP$NP.cm. #前32个恰好是patient1-32的ProximalEnd样本
pd$distance2 <- as.numeric(pd$distance2)
pd$patient <- as.factor(pd$patient)
学习线性混合效应模型
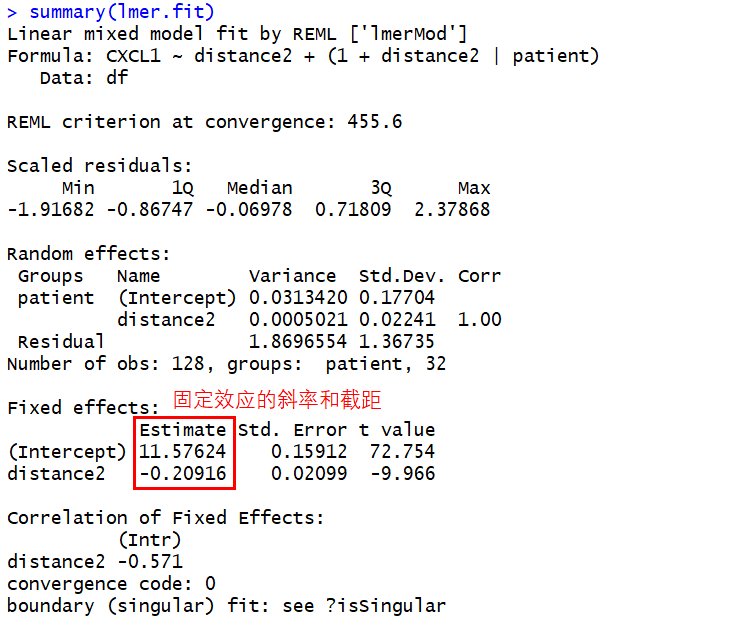
先以基因“CXCL1”的表达量为例,了解线性混合效应模型。
线性混合效应模型包括2个部分:
- 固定效应:被研究的变量,本例中是“距离”
- 随机效应:希望被控制的影响因素,本例中是“患者”
常用的随机效应公式:
(1 | g) :斜率固定,截距变化
x + (x | g) :斜率和截距都变化
#以CXCL1为例
df <- data.frame(CXCL1=expr["CXCL1",],pd[,c(5,7,8)])
library(lme4)
lmer.fit <- lmer(CXCL1~distance2+(1+distance2|patient),data = df)
summary(lmer.fit)
查看随机效应:
# 画图查看随机效应
library(sjPlot)
plot_model(lmer.fit, type = "re", show.values = TRUE)
#查看每个患者分别计算的截距和斜率
coefficients(lmer.fit)
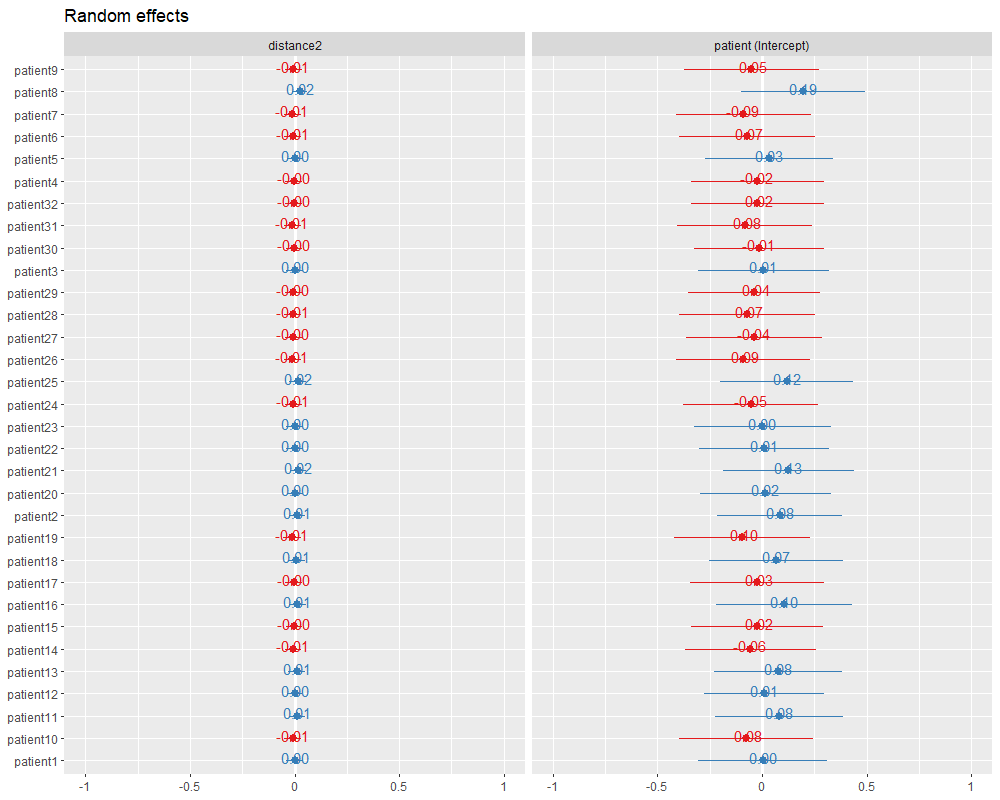
图中2列分别是每个患者的斜率和截距相对于拟合模型的相对差异,例如patient9的斜率为-0.02916-0.01=-0.21916,截距为11.57624-0.05=11.52624。
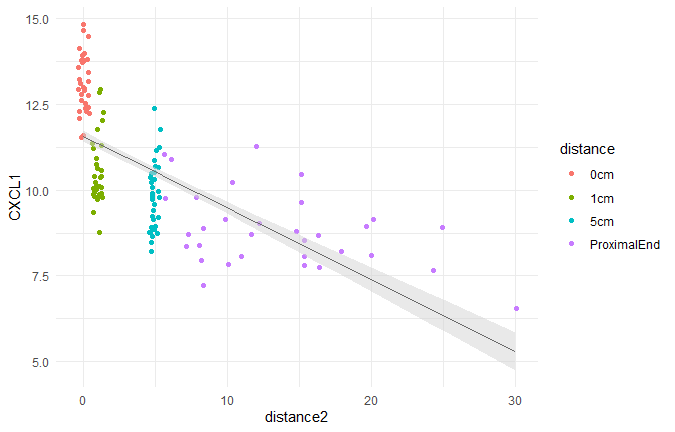
画图查看构建的模型
library(ggeffects)
pred.mm <- ggpredict(lmer.fit, terms = c("distance2"))
ggplot(pred.mm) +
geom_point(data = df,aes(x = distance2, y = CXCL1, colour = distance),position = "jitter") +
geom_line(aes(x = x, y = predicted)) + # slope
geom_ribbon(aes(x = x, ymin = predicted - std.error, ymax = predicted + std.error),
fill = "lightgrey", alpha = 0.5) + # error band
theme_minimal()
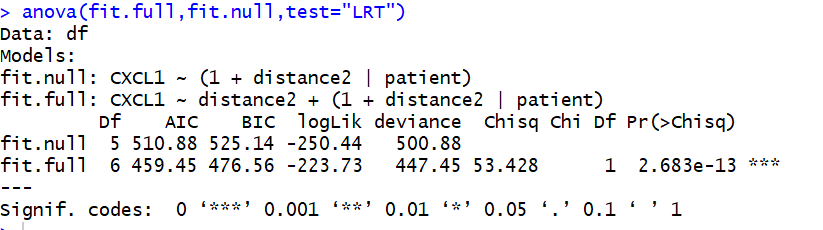
检验固定效应是否显著:比较模型与去除固定效应模型的差异,若2个模型差异显著,则认为固定效应存在。
注意:REML(residualmaximum likelihood),即残差最大似然,默认固定效应是正确的,lmer建模时默认REML=TRUE,所以在判断固定效应是否显著时,要将其设为FALSE,使用ML(最大似然)建模。
#固定效应是否显著
fit.full <- lmer(CXCL1~distance2+(1+distance2|patient),data = df,REML = F)
fit.null <- lmer(CXCL1~(1+distance2|patient),data = df,REML = F)
anova(fit.full,fit.null,test="LRT") #LRT代表极大似然检验
计算所有基因与距离的相关性
## 批量计算
coeff <- list()
pval <- list()
for(i in 1:nrow(expr)){
#i=1
lmer.fit <- lmer(expr[i,]~pd$distance2+(pd$distance2|pd$patient))
coeff[[i]] <- summary(lmer.fit)$coefficients[2,1]
fit.full <- lmer(expr[i,]~pd$distance2+(pd$distance2|pd$patient),REML = F)
fit.null <- lmer(expr[i,]~(pd$distance2|pd$patient),REML = F)
pval[[i]] <- anova(fit.full,fit.null,test="LRT")$`Pr(>Chisq)`[2]
}
lmer.res <- data.frame(gene=rownames(expr),
coeff=unlist(coeff),
pval=unlist(pval),
stringsAsFactors = F)
#去掉重复的基因名
lmer.res <- lmer.res[order(lmer.res$gene,lmer.res$pval),]
lmer.res <- lmer.res[!duplicated(lmer.res$gene),]
#校正FDR
lmer.res$FDR <- p.adjust(lmer.res$pval,method = "bonferroni")
save(lmer.res,file = "./Rdata/lmer_result.Rdata")
与文章结果比较
load("./Rdata/lmer_result.Rdata")
# 按文章的FDR cutoff过滤结果
diff <- lmer.res[lmer.res$FDR < 0.0001,]
dim(diff) #611个基因
table(diff$coeff>0) #上调204个,下调407个
# 读入原文结果(从文章的补充数据下载)
library(readxl)
up_df <- read_excel("./raw-data/oncotarget-08-61107-s002.xls", skip = 1)
down_df <- read_excel("./raw-data/oncotarget-08-61107-s003.xls", skip = 1)
df_paper <- rbind(up_df,down_df)
length(intersect(diff$gene,df_paper$GeneSymbol)) #交集580个
#韦恩图看交集
require("VennDiagram")
VENN.LIST=list(paper=df_paper$GeneSymbol,my_res=diff$gene)
venn.plot <- venn.diagram(VENN.LIST , NULL,
fill=c("#F4E99A", "#95D6F4"),
alpha=c(0.7,0.7), cex = 1.5,
cat.fontface=4)
grid.draw(venn.plot)
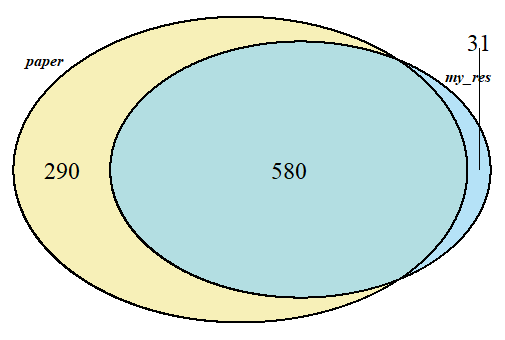
我一共得到611个距离相关基因,原文是870个,交集580个。简单看了一下,系数和p值与原文都是类似的,差异主要来自FDR的校正值,应该是因为我在校正时用的最严格的bonferroni方法,得到的基因数较少。
综上,这个线性混合效应模型的构建应该是没有问题的,剩下的就是基因集的下游分析,这里就不再赘述了,需要的小伙伴可以参见往期教程:
- 1. 公共数据辅助乳腺癌的免疫治疗机制研究
- 2. 有生物学意义的复杂热图
- 3. 干扰MYC‑WWP1通路重新激活PTEN的抑癌活性——3步搞定GSEA分 析
- 4. 按基因在染色体上的顺序画差异甲基化热图
- 5. 热图、⻙恩图、GO富集分析图(有了转录组数据不知道该怎么写⽂ 章,看我就对了!)
- 6. 纯R代码实现ssGSEA算法评估肿瘤免疫浸润程度
- 7. 肿瘤异质性+免疫浸润细胞数据挖掘(可能是Y简单的3分⽂章了)
- 8. ArrayExpress数据库的基因芯⽚原始数据处理,3D主成分图及聚类热 图
- 9. 学徒数据挖掘第⼆期汇总之多分组基因注释代码⼤放送
- 10. TCGA数据辅助甲基化区域的功能研究
- 11. 你确定你的差异基因找对了吗?
- 12. 看nature⽂章是如何设计和使⽤普通转录组数据
- 13. 不⼀定正确的多分组差异分析结果热图展现
- 14. 如果传统bulk转录组数据队列⾜够⼤也可以使⽤单细胞流程
- 15. 最简单的芯⽚挖掘也会出错(菜⻦团周⼀数据挖掘专栏第?期)
- 16. 乳腺癌的IHC分类和PAM50分型的差异情况
- 17. 你要挖的公共数据集作者上传了错误的表达矩阵肿么办(如何让⾼⼿ ⼼⽢情愿的帮你呢?)