昨天我在单细胞天地讲解了使用monocle2进行拟时序分析的方法,基本上跟着我的代码走一波就可以学会了,当然具体参数理解需要自行发力哦,见:使用monocle做拟时序分析(单细胞谱系发育) 用法只是最基础的知识而已,更多的时候,我们需要活学活用,比如课程学员提到的问题,就是因为做不到活学活用,他想知道下面的拟时序分析的热图提取基因,学员把基因按照发育顺序绘制了热图,而这些基因被他分成了3组,想拿基因去做GO/KEGG等数据库进行功能注释,不知道如何获取基因名字。
我这里不能拿学员真实项目数据来演示,所以还是用我们的老朋友,拿scRNAseq包的表达矩阵测试,见:使用monocle做拟时序分析(单细胞谱系发育)
首先根据细胞发育谱系来绘制热图
因为前面的教程 使用monocle做拟时序分析(单细胞谱系发育) 我们已经把细胞发育情况做出来了,就是NPC细胞跟另外3种细胞从生理上就不一样,所以是单独的发育轨迹,而 “GW16” and “GW21” ,“GW21+3” 这种孕期细胞,就可以很清晰的看到时间被反映在我们的拟时序分析结果了。
大家可以重温教程 使用monocle做拟时序分析(单细胞谱系发育) 里面的4个绘图代码:
plot_cell_trajectory(cds, color_by = "Biological_Condition")
# 可以很明显看到细胞的发育轨迹
plot_cell_trajectory(cds, color_by = "State")
plot_cell_trajectory(cds, color_by = "Pseudotime")
plot_cell_trajectory(cds, color_by = "State") +
facet_wrap(~State, nrow = 1)
我们前面构建细胞发育谱系,使用的是不同Biological_Condition的细胞类型之间使用monocle找到的两千多个基因。
简单的一个函数就可以绘制热图:
plot_pseudotime_heatmap(cds[ordering_genes,],
num_clusters = 3,
cores = 1,
show_rownames = T)
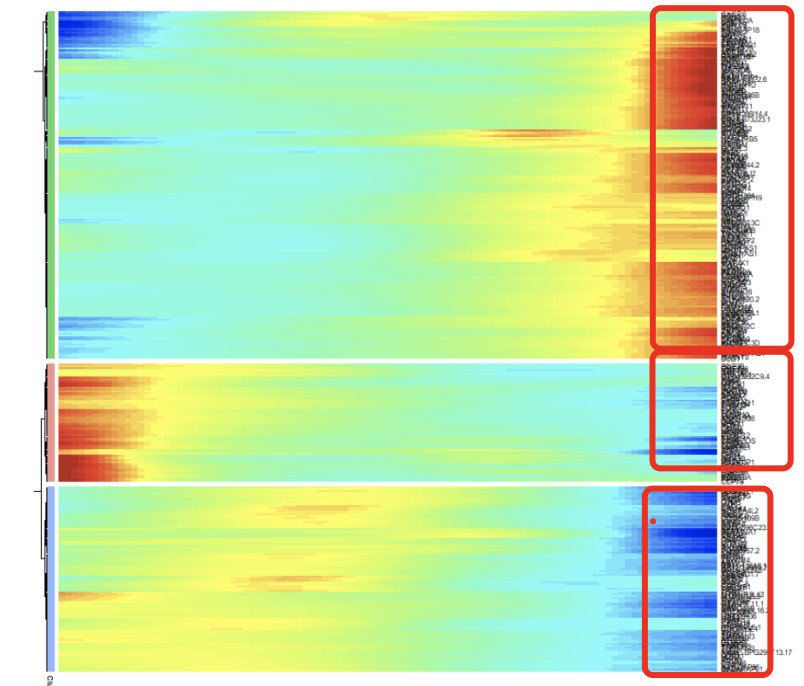
可以看到, 这个图就和学员求助的图一模一样啦,因为基因数量两千多个,所以画出来肯定是看不清晰的啦。
既然基因被分成了3组,想拿基因去做GO/KEGG等数据库进行功能注释,就需要获取基因名字。
这个做不出来,不怪学员,因为正常人很难想到,这个绘图函数其实是可以调整返回数据对象的,而且官网例子也没有提到。
需要看函数的帮助文档,如下:
hclust_method
The method used by pheatmap to perform hirearchical clustering of the rows.
hclust_method = "ward.D2"
return_heatmap
Whether to return the pheatmap object to the user.
很明显,这个函数其实就是pheatmap的一个包装罢了,本质上也是调用 hclust 而已,使用的是ward.D2距离。
然后解析热图函数返回对象
根据帮助文档,我们修改参数,这样monocle的plot_pseudotime_heatmap函数就有返回值了,是一个对象。
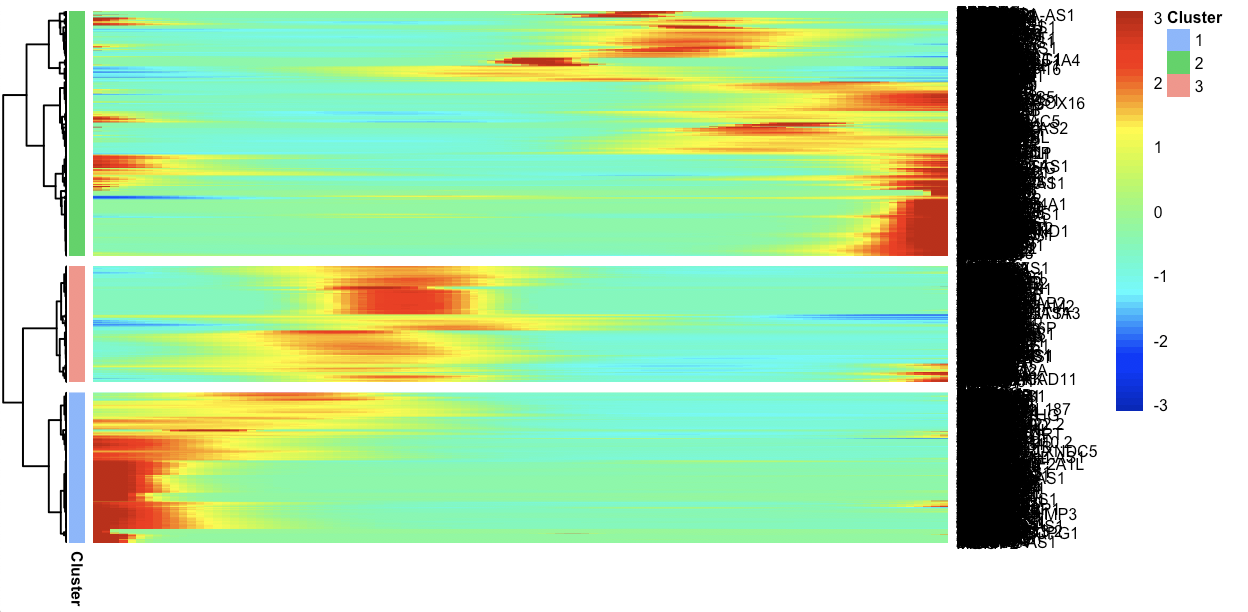
p=plot_pseudotime_heatmap(cds[ordering_genes,],
num_clusters = 3,
cores = 1,return_heatmap=T,
show_rownames = T)
从pheatmap的对象里面提取基因名字就很简单了,就是在p$tree_row里面
> p$tree_row
Call:
hclust(d = d, method = method)
Cluster method : ward.D2
Number of objects: 2200
就可以拿到基因名对应的cluster啦,代码如下:
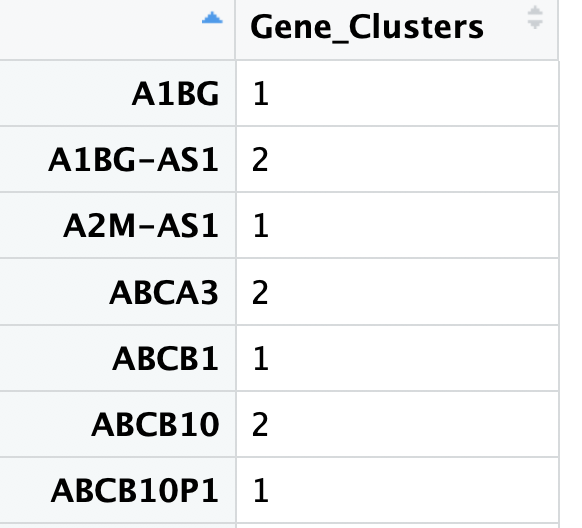
clusters <- cutree(p$tree_row, k = 3)
clustering <- data.frame(clusters)
clustering[,1] <- as.character(clustering[,1])
colnames(clustering) <- "Gene_Clusters"
table(clustering)
实际上学员提问方式本身就是有问题的
因为学员直接就丢出这个热图,然后咨询如何在图片里面提取基因名字,所以大家只能是问是pdf还是png的图片呢?是不是可以AI或者PS解析它,拿到基因名字呢?
如果学员是直接问:使用monocle的plot_pseudotime_heatmap函数绘制的热图里面的基因聚集成为3类,该如何提取基因名字,其实就很简单了。
这个答疑虽然是给基础课程的学员,但实际上这个内容在单细胞转录组的进阶课程,如果学了就不需要提问了。
文末友情宣传
强烈建议你推荐给身边的博士后以及年轻生物学PI,多一点数据认知,让他们的科研上一个台阶:
- 全国巡讲全球听(买一得五) ,你的生物信息学入门课
- 生信技能树的2019年终总结 ,你的生物信息学成长宝藏
- 2020学习主旋律,B站74小时免费教学视频为你领路