就是STAR-fusion啦,它可以直接基于STAR比对好的bam文件来做分析,而大多数其它融合基因查找工具,需要从fastq文件开始,不太方便。之前我在生信技能树公众号介绍过它,那个时候发表该工具的文章是:STAR-Fusion: Fast and Accurate Fusion Transcript Detection from RNA-Seq 在biorxiv预印本:
两年前我在生信技能树就写过它的教程:使用STAR-fusion来对转录组数据找融合基因 也强调大家自行读STAR-Fusion详细说明:https://github.com/STAR-Fusion/STAR-Fusion/wiki 今天(2019-10-29 )翻它的主页发现上面写着已经正式发表了:
还设计了一个好看(简洁)的logo,我留意到比作者少了很多:

而且才意识到,居然是 Aviv Regev 实验室出品的工具!如果生信技能树的粉丝们有在 Aviv Regev 实验室做科研的(博士,博士后均可),希望可以联系我一下,认识一下。
再次回顾STAR-fusion对RNA-seq测序数据找融合基因的流程
生物信息学鉴定融合转录本的方法一般有两种:
- ①将RNA-seq数据与Reference genome做alignment,鉴别可能发生重排的基因;
- ②先直接将reads装配成更长的转录本序列,再鉴别与重排序列一致的融合转录本。
而我们选择的Broad Institute的 Brian J. Haas 和冷泉港实验室(CSHL)的 Alex Dobin 等人开发的工具STAR-Fusion,其工作原理分为三步:
- ①先将reads通过STAR比对到参考基因组,筛选出split和discordant reads作为候选的融合基因序列;
- ②将候选融合基因序列与参考基因序列进行比对,根据overlaps预测出融合基因;
- ③对预测结果做过滤,去除假阳性结果。
首先对RNA-seq测序数据进行star的two-pass比对:
批量运行,其中star_index 需要指定自己的star索引路径文件夹,而bin_star需要指定自己的star软件的可执行程序路径哦。
核心代码是:
start=$(date +%s.%N)
echo star start `date`
$bin_star --runThreadN 4 --genomeDir $star_index \
--twopassMode Basic --outReadsUnmapped None --chimSegmentMin 12 \
--alignIntronMax 100000 --chimSegmentReadGapMax parameter 3 \
--alignSJstitchMismatchNmax 5 -1 5 5 \
--readFilesCommand zcat --readFilesIn $fq1 $fq2 --outFileNamePrefix ${sample}_
mv ${sample}_Aligned.out.sam $sample.sam
$bin_samtools sort -o $sample.bam $sample.sam
$bin_samtools index $sample.bam
$bin_samtools flagstat $sample.bam > $sample.flagstat
touch ok.star.$sample.status
rm $sample.sam
echo star end `date`
dur=$(echo "$(date +%s.%N) - $start" | bc)
printf "Execution time for star : %.6f seconds" $dur
实际上就是一行命令在运行比对过程,但是呢,参数太多了,调起来很麻烦,通常如果不理解的话就不建议修改参数。 有一个链接需要注意:https://github.com/STAR-Fusion/STAR-Fusion/issues/104
值得注意的是,star这个比对软件,也需要选择比较新的版本哦:Please be sure to use the STAR aligner with min version: 2.7.0f 毕竟我上次写教程介绍它的时候是2017年。软件需要最新版,下载的数据库也需要最新版!
比对本来是为了得到bam文件,但是我们这个流程是为了 得到’Chimeric.out.junction’这个文件,供STAR-fusion使用。最新版软件,关于得到’Chimeric.out.junction’这个文件,有一个参数:chimOutJunctionFormat 需要注意。
然后使用conda安装STAR-fusion
因为这个工具大量依赖perl模块,一般人不会这个语言,弄起来也麻烦,所以推荐conda一键式安装
一步法安装代码如下:
conda install -c bioconda star-fusion
可以看到下面那么多的perl模块都会被自动安装:
一般来说,安装成功后,可以使用perl代码检测一下:
perl -e 'use Set::IntervalTree'
# 值得注意的是
STAR-Fusion
#软件命令是 STAR-Fusion
如果没有明显的报错,说明你安装成功啦。如果你不想使用conda一步到位的安装,那么你会面临perl模块的巨坑:
A typical perl module installation may involve:
perl -MCPAN -e shell
install DB_File
install URI::Escape
install Set::IntervalTree
install Carp::Assert
install JSON::XS
install PerlIO::gzip
所以自己衡量一下时间和精力哦,不要逞强!
通常是软件版本问题
比如star版本就很奇怪,在GitHub是最新的:
# Get latest STAR source from releases
wget https://github.com/alexdobin/STAR/archive/2.7.3a.tar.gz
tar -xzf 2.7.3a.tar.gz
cd STAR-2.7.3a
# Alternatively, get STAR source using git
git clone https://github.com/alexdobin/STAR.git
但是conda安装的是2.7.0d , 就有可能报错:
qiEXITING because of FATAL ERROR: Genome version: 2.7.1a is INCOMPATIBLE with running STAR version: 2.7.0f
SOLUTION: please re-generate genome from scratch with running version of STAR, or with version: 2.7.0d
's
Oct 29 20:10:37 ...... FATAL ERROR, exiting
其实conda可以安装指定版本的软件哦!
而且上面的那个报错其实也不是字面上的意思。
需要下载指定参考基因组的数据库文件供STAR-fusion使用
这个需要考验大家网速了,其wiki说明:https://github.com/STAR-Fusion/STAR-Fusion/wiki 写的很清楚。需要注意的是保证参考基因组的一致性哦!
如果你有自己的star软件索引,就下载2.9G的,如果没有,就下载29G的,如果你是人类,下载37和38均可,取决于你对参考基因组的熟悉程度! https://data.broadinstitute.org/Trinity/CTAT_RESOURCE_LIB/ 如果你不熟悉,最好是选择最新版,不要选择我下面截图的那些!!!
反正我下载的是:GRCh38_gencode_v31_CTAT_lib_Oct012019.plug-n-play.tar.gz ,足足31个G!
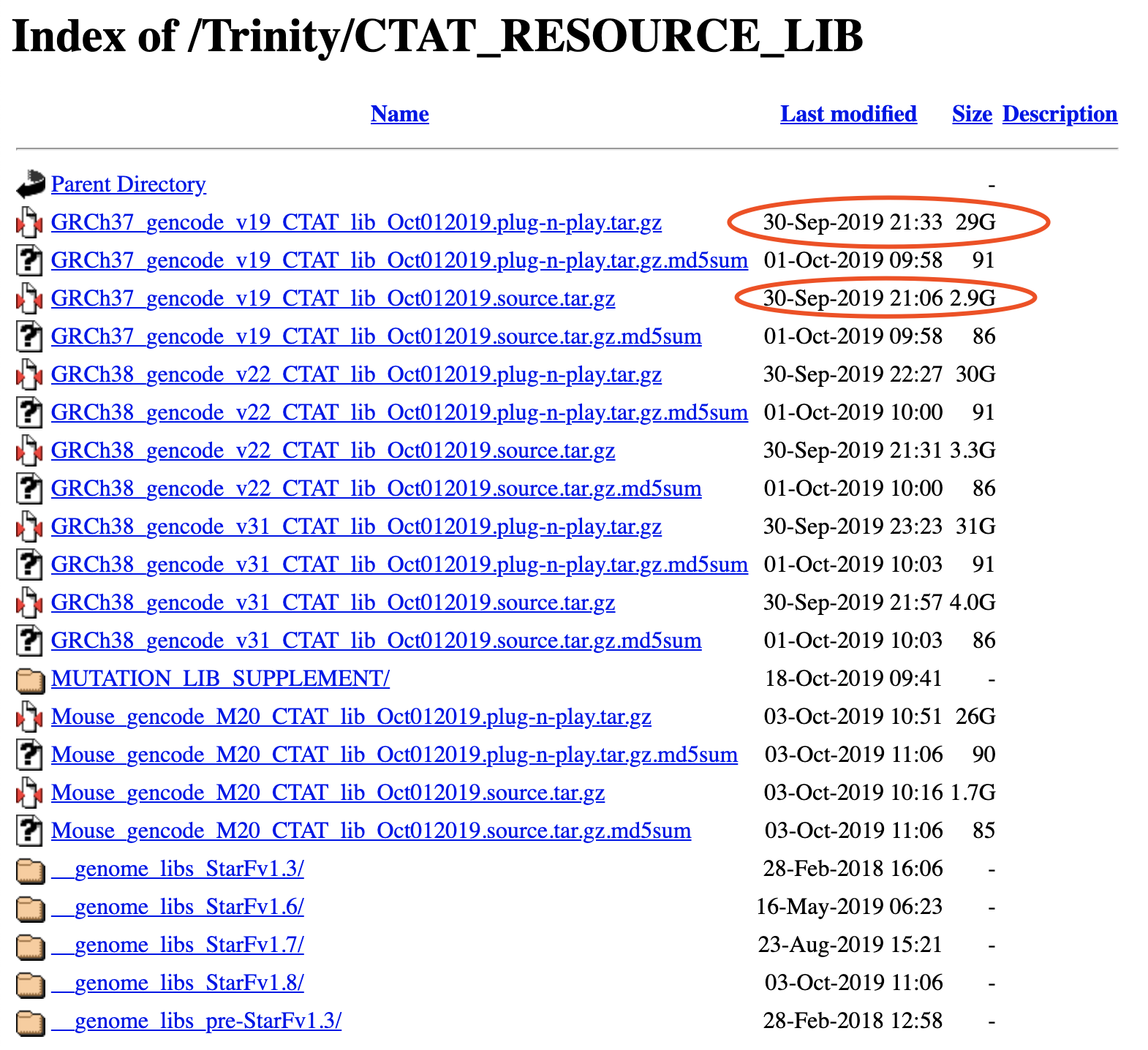
而且需要下载 ; be sure to use a more modern version of the companion CTAT_GENOME_LIB 如果你报错的话,通常就是这些问题。
比如旧版的如下:
新版数据库如下:
运行STAR-fusion
同样也是批量运行:
lib='/home/yb77613/biosoft/starFusion/db/GRCh38_gencode_v31_CTAT_lib_Oct012019.plug-n-play/ctat_genome_lib_build_dir/'
# Please be sure to use the STAR aligner with min version: 2.7.0f
ls ../alignment/*.junction |while read id
do
file=$(basename $id )
sample=${file%%.*}
~/biosoft/starFusion/STAR-Fusion/STAR-Fusion --genome_lib_dir $lib -J $id --output_dir $sample
done
或者单独运行:
lib='/home/yb77613/biosoft/starFusion/db/GRCh38_gencode_v31_CTAT_lib_Oct012019.plug-n-play/ctat_genome_lib_build_dir/'
STAR-Fusion --genome_lib_dir $lib -J Lib_FUSCCTNBC001_Chimeric.out.junction --output_dir s1
实际上,是非常多命令的组合,只不过你自己只需要运行一句话命令即可,比如:
CMD: mkdir -p /home/yb77613/data/public/tnbc/RNA-seq/star/s1
CMD: mkdir -p /home/yb77613/data/public/tnbc/RNA-seq/star/s1/_starF_checkpoints
CMD: mkdir -p /home/yb77613/data/public/tnbc/RNA-seq/star/s1/star-fusion.preliminary
-sample contains 41047586
* Running CMD: /home/yb77613/miniconda3/envs/rna/lib/STAR-Fusion/util/STAR-Fusion.map_chimeric_reads_to_genes --genome_lib_dir /home/yb77613/biosoft/starFusion/db/GRCh38_gencode_v31_CTAT_lib_Oct012019.plug-n-play/ctat_genome_lib_build_dir/ -J /home/yb77613/data/public/tnbc/RNA-seq/star/Lib_FUSCCTNBC001_Chimeric.out.junction > /home/yb77613/data/public/tnbc/RNA-seq/star/s1/star-fusion.preliminary/star-fusion.junction_breakpts_to_genes.txt
具体大家可以去查看log日志哈。
输出文件的解读
其实说明书也写的很清楚了,主要就是看JunctionReads列,看看有多少条序列支持这样的融合,然后也会列出融合基因的断点出左右两个基因的衔接位置的碱基等等。
#FusionName JunctionReadCount SpanningFragCount SpliceType LeftGene LeftBreakpoint RightGene RightBreakpoint LargeAnchorSupport FFPM LeftBreakDinuc LeftBreakEntropy RightBreakDinuc RightBreakEntropy annots
IGH-@--MALAT1 4 16 INCL_NON_REF_SPLICE IGH-@^IGH-.g@ chr14:105708080:+ MALAT1^ENSG00000251562.8 chr11:65499045:+ YES_LDAS 0.4385 TG 1.7232 GT 1.8892 ["TCGA_StarF2019","INTERCHROMOSOMAL[chr14--chr11]"]
IGH-@-ext--MALAT1 4 16 INCL_NON_REF_SPLICE IGH-@-ext^IGH-.g@-ext chr14:105708080:+ MALAT1^ENSG00000251562.8 chr11:65499045:+ YES_LDAS 0.4385 TG 1.7232 GT 1.8892 ["INTERCHROMOSOMAL[chr14--chr11]"]
IGH-@--KRT19 5 9 INCL_NON_REF_SPLICE IGH-@^IGH-.g@ chr14:105742042:+ KRT19^ENSG00000171345.13 chr17:41528296:- YES_LDAS 0.3069 GC 1.6729 CG 1.9899 ["TCGA_StarF2019","INTERCHROMOSOMAL[chr14--chr17]"]
IGH-@-ext--KRT19 5 9 INCL_NON_REF_SPLICE IGH-@-ext^IGH-.g@-ext chr14:105742042:+ KRT19^ENSG00000171345.13 chr17:41528296:- YES_LDAS 0.3069 GC 1.6729 CG 1.9899 ["INTERCHROMOSOMAL[chr14--chr17]"]
AFF4--MAPK8 2 10 ONLY_REF_SPLICE AFF4^ENSG00000072364.13 chr5:132937067:- MAPK8^ENSG00000107643.16 chr10:48401612:+ YES_LDAS 0.2632 GT 1.9219 AG 1.7232 ["INTERCHROMOSOMAL[chr5--chr10]"]
AFF4--MAPK8 1 10 ONLY_REF_SPLICE AFF4^ENSG00000072364.13 chr5:132937067:- MAPK8^ENSG00000107643.16 chr10:48325897:+ YES_LDAS 0.2412 GT 1.9219 AG 1.9086 ["INTERCHROMOSOMAL[chr5--chr10]"]
RRM2B--AC016074.2 2 4 ONLY_REF_SPLICE RRM2B^ENSG00000048392.11 chr8:102224046:- AC016074.2^ENSG00000286122.1 chr8:125650233:+ YES_LDAS 0.1316 GT 1.8323 AG 1.7465 ["INTRACHROMOSOMAL[chr8:23.41Mb]"]
然后新版的STAR-fusion增加了一个过滤的功能,主要是过滤掉那些线粒体基因融合好HLA相关基因融合,说明书一直提到了:Red Herrings: Fusion pairs that may not be relevant to cancer, and potential false positives. 主要是因为这个软件开发就是为癌症研究服务的。
如果你确实不需要这个默认的过滤功能,就需要修改参数啦。
STAR-fusion还有两个好哥们
If you plan on using the included FusionInspector for ‘inspect’ or ‘validate’ modes, please install the FusionInspector dependencies. 主要是可视化你的融合事件,通常是基于IGV。还有个是 FusionAnnotator 主要是基于癌症数据库,来为你的融合事件推荐注释。
后面我们根据粉丝的学习情况来看看是否需要加餐继续介绍