因为10X仪器的商业化成功,目前大家的单细胞转录组课题基本上都是10X数据,所以我在单细胞天地分享了一系列相关教程,希望可以接地气的帮助大家,如下:
其中在上面的两个样品的10x单细胞转录组数据分析策略 教程里面,我委婉的指出来了,那个文章对两个样本的10X单细胞转录组数据的整合是有问题的,现在我们就来慢慢讨论一下这个问题。
首先批次效应还是真实生物学差异是可以区分的
在教程:使用seurat3的merge功能整合8个10X单细胞转录组样本 和 seurat3的merge功能和cellranger的aggr整合多个10X单细胞转录组对比 我其实展示了如果10X的样本效应被去除,应该是什么样的效果,如下:

同样的发育时期的2个样本,细胞亚群是类似的,但是不同发育时期的细胞亚群差异很大,所以可以初步认为,同样的发育时期的不同样本之间是没有批次效应的,而不同发育时期的差异应该是真实的生物学差异,并不是批次效应。
如果只有一个样本
但是上面的两个样品的10x单细胞转录组数据分析策略 的文章里面,作者仅仅是做了一个野生型和一个突变的小鼠,所以它们两个样本的差异,就必然会混入批次效应和真实生物学差异,需要仔细探索。
首先下载作者文章里面公布的原始数据
因为这个数据集的文章作者使用的是cellranger流程,而且我们在单细胞天地多次分享过流程笔记,大家可以自行前往学习,如下:
- 单细胞实战(一)数据下载
- 单细胞实战(二) cell ranger使用前注意事项
- 单细胞实战(三) Cell Ranger使用初探
- 单细胞实战(四) Cell Ranger流程概览
- 单细胞实战(五) 理解cellranger count的结果
得到表达矩阵的报表如下:
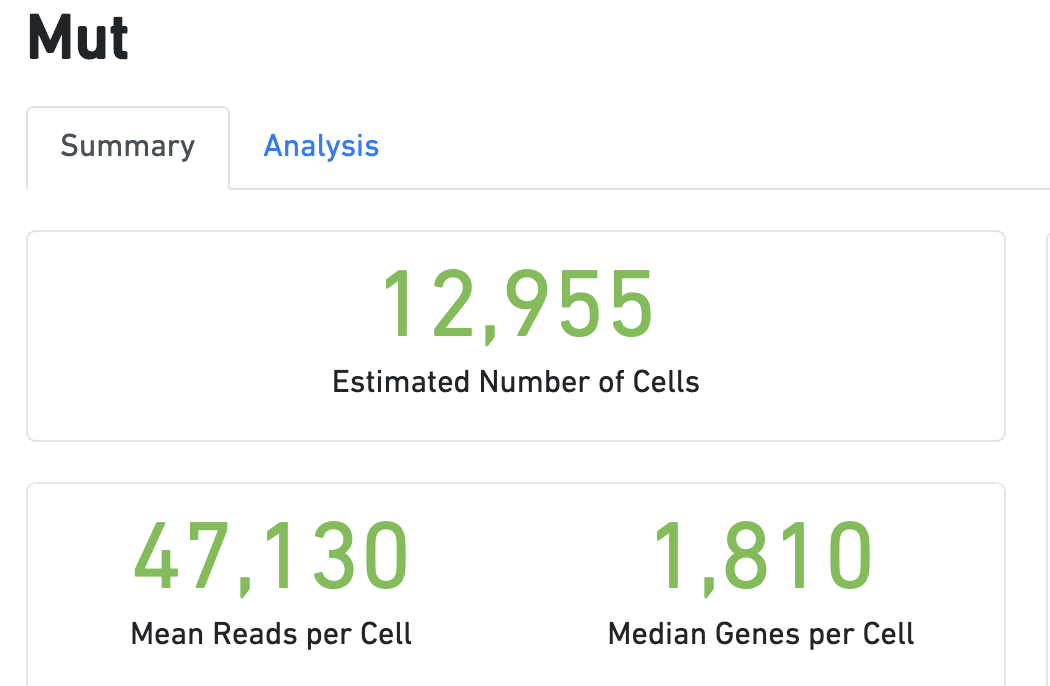
跟文章描述的差不多。
然后样本合并走seurat的merge流程
就是前面我们的教程:使用seurat3的merge功能整合8个10X单细胞转录组样本 的代码:
rm(list=ls())
options(stringsAsFactors = F)
library(Seurat)
sce1 <- CreateSeuratObject(Read10X('../10x-results/WT/'),
"wt")
sce2 <- CreateSeuratObject(Read10X('../10x-results/mut/'),
"mut")
sce<- merge(sce1,y = c(sce2),
add.cell.ids = c('wt','mut'),
project = "mouse")
sce
head(sce@meta.data)
GetAssayData(sce,'counts')
sce[["percent.mt"]] <- PercentageFeatureSet(sce, pattern = "^mt-")
sce[["percent.ribo"]] <- PercentageFeatureSet(sce, pattern = "^Rp[sl]")
sce <- subset(sce,
subset = nFeature_RNA > 200 & nFeature_RNA < 5000 & percent.mt < 10)
sce <- NormalizeData(sce, normalization.method = "LogNormalize", scale.factor = 10000)
sce <- FindVariableFeatures(sce, selection.method = "vst", nfeatures = 2000)
plot1 <- VariableFeaturePlot(sce)
plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE)
CombinePlots(plots = list(plot1, plot2),legend = 'none')
# 步骤 ScaleData 的耗时取决于电脑系统配置(保守估计大于一分钟)
sce <- ScaleData(sce)
sce <- RunPCA(object = sce, pc.genes = VariableFeatures(sce))
VizDimLoadings(sce, dims = 1:2, reduction = "pca")
DimHeatmap(sce, dims = 1:12, cells = 100, balanced = TRUE)
ElbowPlot(sce)
sce <- FindNeighbors(sce, dims = 1:15)
sce <- FindClusters(sce, resolution = 0.2)
table(sce@meta.data$RNA_snn_res.0.2)
table(sce@meta.data$RNA_snn_res.0.2,sce@meta.data$orig.ident)
set.seed(123)
sce <- RunTSNE(object = sce, dims = 1:15, do.fast = TRUE)
DimPlot(sce,reduction = "tsne",label=T)
DimPlot(sce,reduction = "tsne",label=T,split.by ='orig.ident')
为了达到作者文章里面的11群细胞,我调整了resolution参数,低到了0.2,哈哈哈
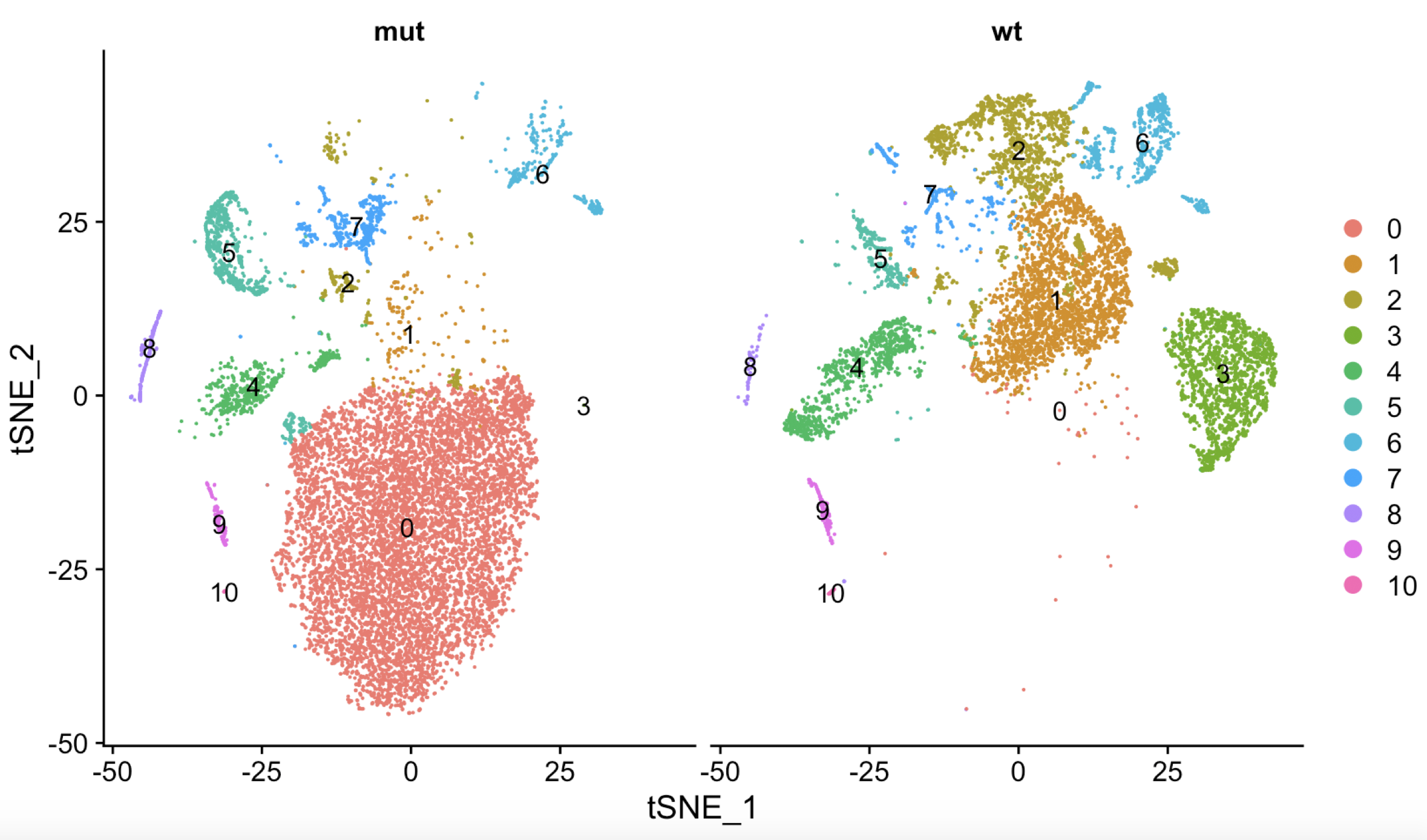
而且可以看到,跟文章的图表几乎是一模一样,在mut的小鼠里面有一群细胞非常突出!
上面演示的seurat包来进行多样本合并代码,需要大家最好是有基础知识,比如听完我的全网第一个单细胞课程(基础)满一千份销量就停止发售 内容,就是一些R包的认知,包括 scater,monocle,Seurat,scran,M3Drop 需要熟练掌握它们的对象,:一些单细胞转录组R包的对象 ,分析流程也大同小异:
- step1: 创建对象
- step2: 质量控制
- step3: 表达量的标准化和归一化
- step4: 去除干扰因素(多个样本整合)
- step5: 判断重要的基因
- step6: 多种降维算法
- step7: 可视化降维结果
- step8: 多种聚类算法
- step9: 聚类后找每个细胞亚群的标志基因
- step10: 继续分类
具体研究差异最大的细胞亚群
因为只有一个样本,所以没办法根据分群结果就判定是生物学差异还是批次效应,需要仔细的去探索具体的每个亚群!
简单整理一下,可以发现mut和wt的小鼠,差异很大的就是0-3这4群细胞啦。

首先查看是否不同亚群细胞有很明显的批次因素混杂,如下,可以看到不同亚群之间的批次因素并不是其生物学差异的混杂(这里,我很难使用文字描述,你需要仔细看上图和下图,尝试理解)
然后需要具体查看细胞的标记基因,那就得使用 FindMarkers 函数啦!
对文章的11个细胞亚群分别找差异基因,然后热图可视化,代码如下:
sce.markers <- FindAllMarkers(object = sce, only.pos = TRUE, min.pct = 0.25,
thresh.use = 0.25)
write.csv(sce.markers,file = 'all_sce.markers.csv')
library(dplyr)
top10 <- sce.markers %>% group_by(cluster) %>% top_n(10, avg_logFC)
DoHeatmap(sce,top10$gene)
出图如下:

因为确实没有这篇文章的生物学背景,我能看出来部分基因其实是有意义的,比如KRT相关基因,CCL相关基因,S100相关基因。
这个时候,可以对每个亚群的标记基因拿去做GO/KEGG数据库注释,这个方法在今天的生信技能树推文有讲解:重复一篇WGCNA分析的文章(代码版) , 在 step5_Annotation_Module_GO_BP:
那么到底是批次效应还是真实生物学差异呢
其实到目前为止的探索,我并不能完全确定,很多粉丝在后台都表明急缺生信工程师加盟课题组分析数据,实际上呢,我们生信工程师何尝不想找一个生物学背景足够强的合作者呢?