前面我在生信技能树推文:你确定你的差异基因找对了吗? 提出了文章的转录组数据的60个样品并没有按照毒品上瘾与否这个表型来区分,而是不同人之间的异质性非常高,这个时候我提出来了一个解决方案,就是理论上就可以把人当做是一个批次效应,使用北京大学李程课题组开发的sva包的combat函数,把这样的效应去除一下,接着再找差异。
当然了,去除批次效应的方法,肯定不止这一个,现在让我们列举并且比较一下吧!
首先载入数据并且包装一个PCA可视化函数
rm(list = ls()) ## 魔幻操作,一键清空~
options(stringsAsFactors = F)
pca_plot = function(dddd,ggggg){
library("FactoMineR")
library("factoextra")
df.pca <- PCA(t(dddd), graph = FALSE)
fviz_pca_ind(df.pca,
#axes = c(2,3),
geom.ind = "point",
col.ind = ggggg ,
addEllipses = TRUE,
legend.title = "Groups"
)
}
# 下面的 step1-output.Rdata 文件,大家可以去学习我的GEO课程,就知道如何制作啦。
load(file = 'step1-output.Rdata')
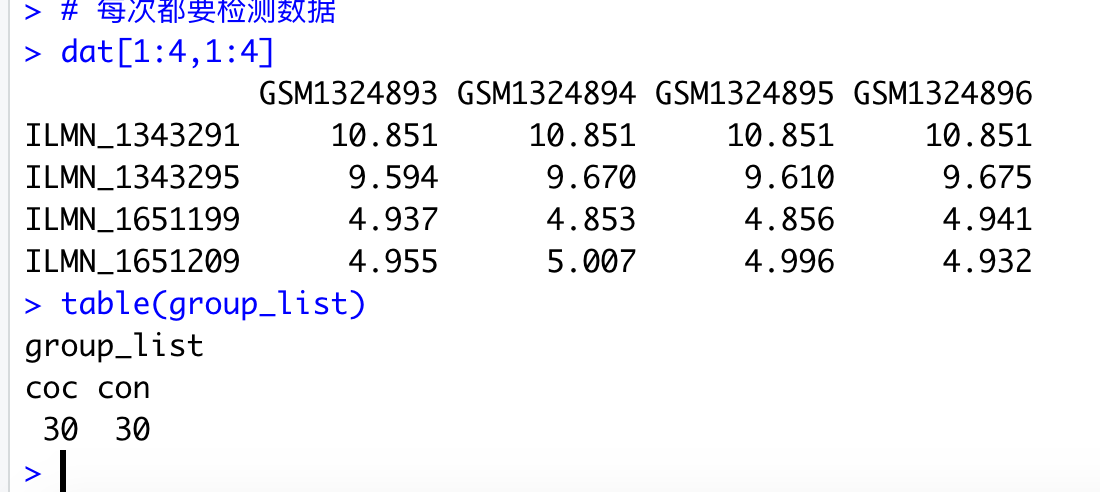
# 每次都要检测数据
dat[1:4,1:4]
table(group_list)
library(limma)
g=factor( group_list )
g
g=relevel(g,'con')
design=model.matrix(~g)
fit=lmFit(dat,design)
fit=eBayes(fit)
options(digits = 4)
topTable(fit,coef=2,adjust='BH')
# 首先是瘾君子与正常人的差异分析
deg1=topTable(fit,coef=2,adjust='BH',number = Inf)
pca_plot(dat,g)
数据如下,是一个表达矩阵和分组信息,我在B站的GEO课程多次讲解了,大家读懂:
使用 limma 的 removeBatchEffect 函数
需要注意的是removeBatchEffect 函数这里表达矩阵和需要被去除的批次效应是必须参数,然后本来的分组也是需要添加进入,这样与真实分组相关的差异就会被保留下来。
load('trait.Rdata')
batch=trait$patient
## 使用 limma 的 removeBatchEffect 函数
dat[1:4,1:4]
ex_b_limma <- removeBatchEffect(dat,
batch = batch,
design = design)
dim(ex_b_limma)
ex_b_limma[1:4,1:4]
fit=lmFit(ex_b_limma,design)
fit=eBayes(fit)
options(digits = 4)
topTable(fit,coef=2,adjust='BH')
deg2=topTable(fit,coef=2,adjust='BH',number = Inf)
pca_plot(ex_b_limma,g)
然后使用 sva 的 ComBat 函数
可能是我没有理解 sva 的 ComBat 函数用法,下面的代码发现并不能把个体差异抹去,无法凸显出毒品成瘾与否两个分组的差异。
## 使用 sva 的 ComBat 函数
library(sva)
table(batch)
table(g)
ex_b_sva = ComBat(dat=as.matrix(dat),
batch=batch
)
ex_b_sva[1:4,1:4]
fit=lmFit(ex_b_sva,design)
fit=eBayes(fit)
options(digits = 4)
topTable(fit,coef=2,adjust='BH')
# 首先是瘾君子与正常人的差异分析
deg3=topTable(fit,coef=2,adjust='BH',number = Inf)
pca_plot(ex_b_sva,g)
比较原始矩阵和去除批次效应后
可以看到,只有 limma 的 removeBatchEffect 函数做到了把矩阵区分成为毒品上瘾与否的截然不同的两个部分。
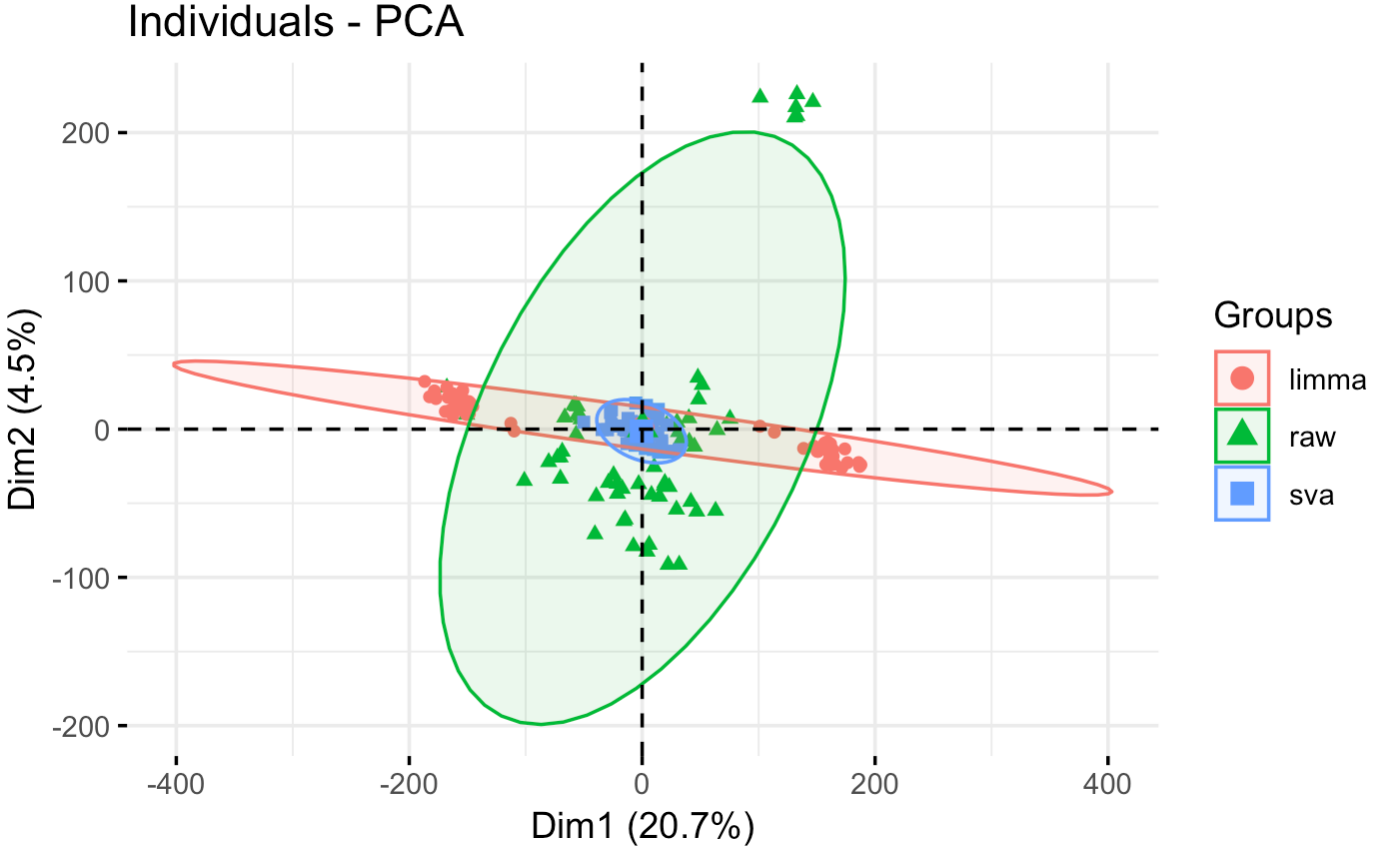
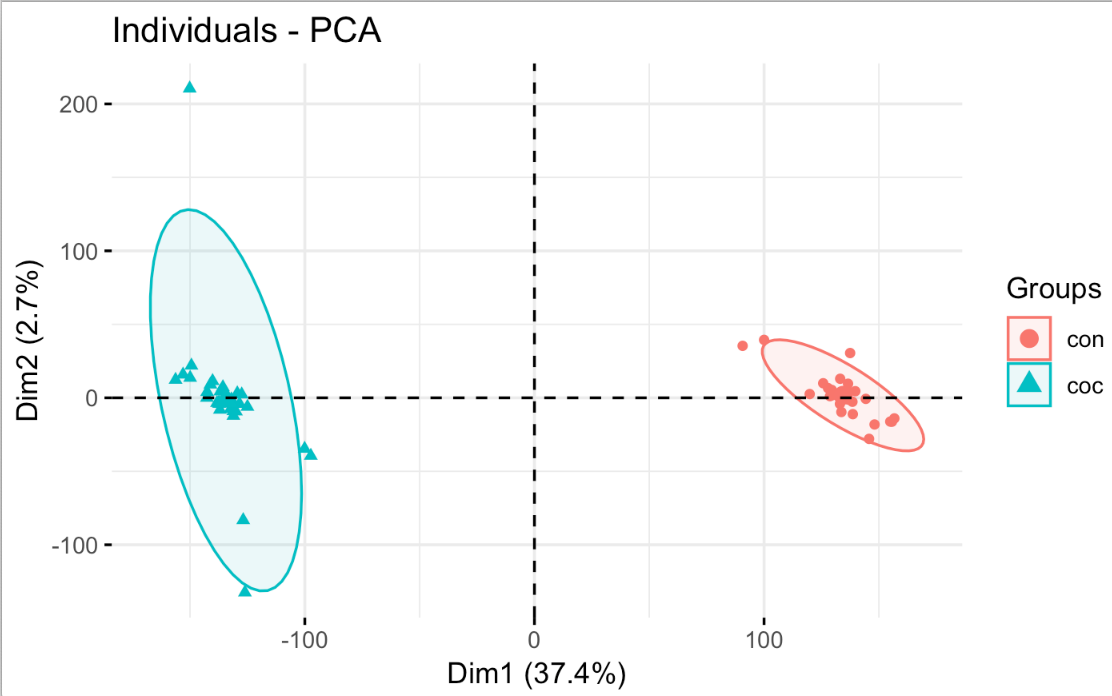
毫无疑问,使用这样的去除了人的效应的表达矩阵后再做差异分析肯定是能找到非常多的有统计学显著效果的基因列表。单独PCA可视化 limma 的 removeBatchEffect 函数后的表达矩阵,如下:
把3次差异分析结果结合起来
ids=rownames(deg1)
combDEG=cbind(deg1[ids,c(1,5)],
deg2[ids,c(1,5)],
deg3[ids,c(1,5)])
cor(combDEG[,c(1,3,5)])
那些之前被人群效应淹没的毒品上瘾相关的基因有:
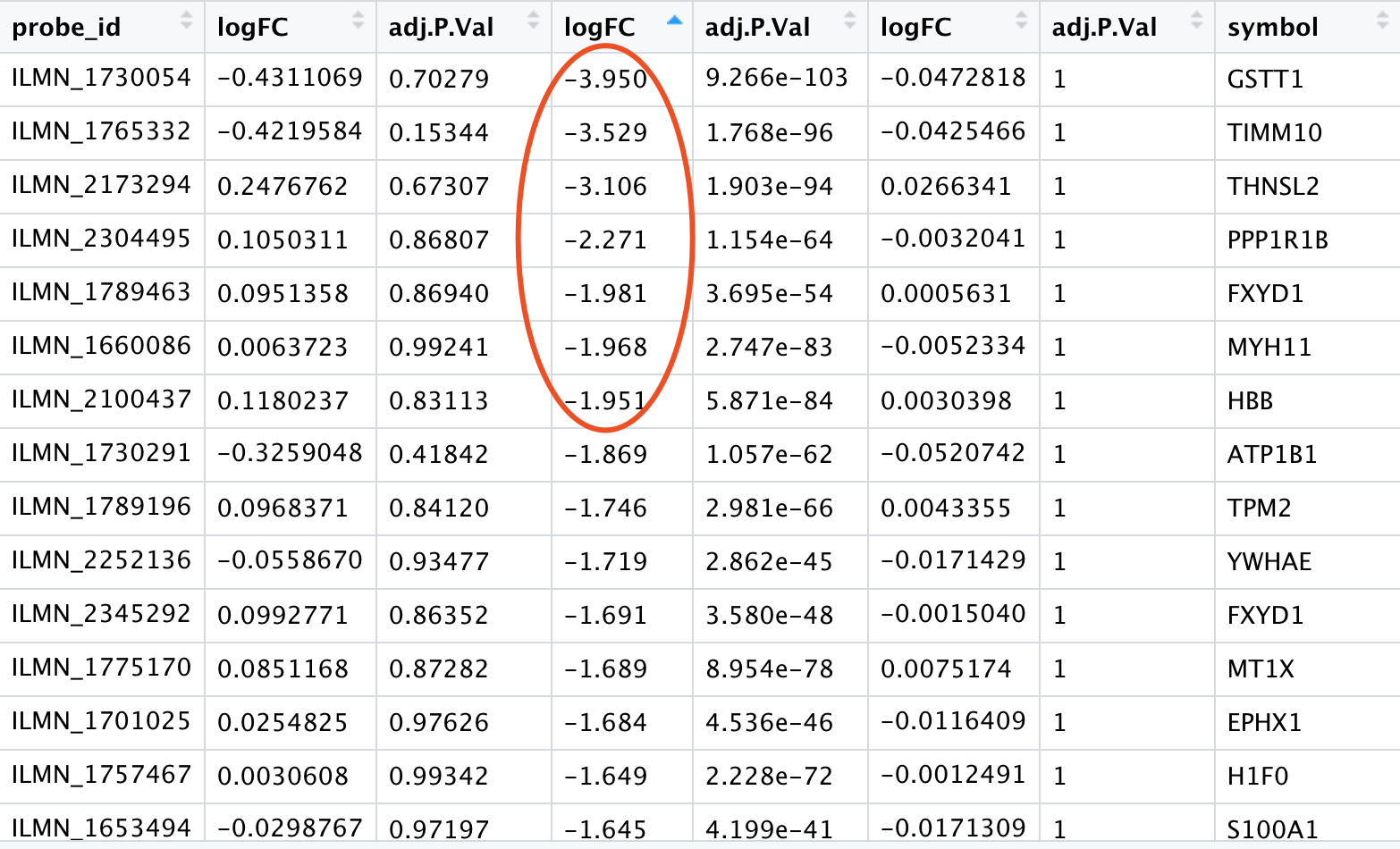
然后我们去可视化这些基因,在两个表达矩阵:
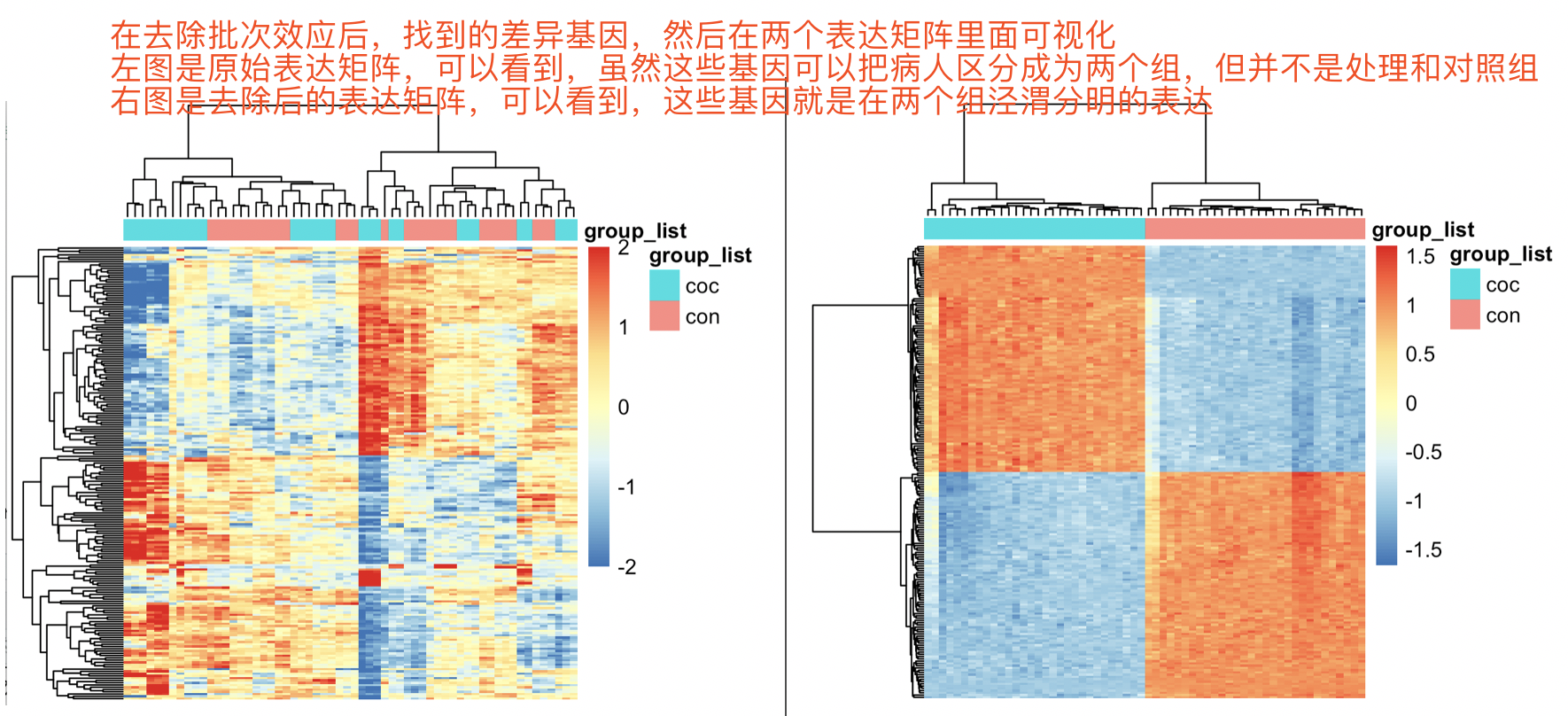
代码很简单:
combDEG$probe_id=rownames(combDEG)
library(illuminaHumanv3.db)
ids=toTable(illuminaHumanv3SYMBOL)
deg=merge(combDEG,ids,by='probe_id')#通过merge函数,由于deg和ids都有probe_id这一列,因此通过'probe_id'合并为新的deg
deg=deg[order(deg[,4]),]
cg=c(head(deg[,1],100),
tail(deg[,1],100))
# ex_b_limma, ex_b_sva, dat
library(pheatmap)
n=t(scale(t(ex_b_limma[cg,])))
n[n>2]=2 #限定上限,使表达量大于2的等于2
n[n< -2]= -2 #限定下限,使表达量小于-2的等于-2
n[1:4,1:4]
load('trait.Rdata')
ac=data.frame(group_list=group_list)
rownames(ac)=colnames(n)
pheatmap(n,show_colnames =F,show_rownames = F,annotation_col = ac)
现在的问题就是,我们定位的这些差异基因,是否在真正的两个组别的差异呢,还是仅仅是因为我们使用了算法抹去个体差异后的产物。