NCBI的blast++软件的使用
目录
一:下载安装该软件
二:准备数据
三:运行命令
四:输出文件解读
正文
一:下载安装该软件
在NCBI的ftp站点里面可以找到blast++的下载链接
wget ftp://ftp.ncbi.nlm.nih.gov/blast/executables/LATEST/ncbi-blast-2.2.30+-x64-linux.tar.gz
我们一般选择适合我们操作系统的二进制版本,解压即可使用
可以把它们添加到PATH,前提是有root权限,或者把该目录添加到PATH也行。
cp * /home/jmzeng/my-bin/bin/
我把my-bin添加到了我的PATH,所以可以直接使用这些程序了
二:准备数据
只需要fasta文件的数据即可,query和target都可以是该fasta文件,可以随便找两个fa文件做测试
三:运行命令
1,建库,用makeblastdb,标准是
makeblastdb -in db.fasta -dbtype prot -parse_seqids -out dbname
具体参数看help里面的,但是我们一般用这几个就够了的
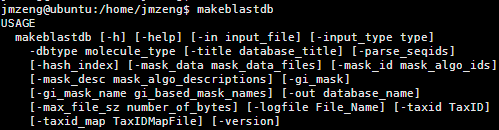
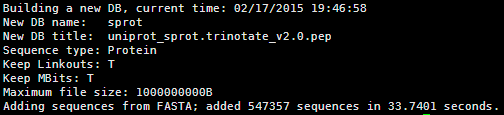
我的例子 :对200M的蛋白文件
makeblastdb -in uniprot_sprot.trinotate_v2.0.pep -dbtype prot -parse_seqids -out sprot
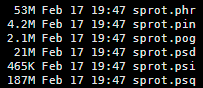
输出的文件如下,基本不需要看,反正调用的时候只用sprot这个
对8G的uniref90,
makeblastdb -in uniprot_uniref90.trinotate_v2.0.pep -dbtype prot -parse_seqids -out uniref90
2,比对分为好几种,blastn, blastp,blastx,tblastn,tblastx
- blastp -query seq.fasta -out seq.blast -db dbname -outfmt 6 -evalue 1e-5 -num_descriptions 10 -num_threads 8
- blastn -query seq.fasta -out seq.blast -db dbname -outfmt 6 -evalue 1e-5 -num_descriptions 10 -num_threads 8
- blastx -query seq.fasta -out seq.blast -db dbname -outfmt 6 -evalue 1e-5 -num_descriptions 10 -num_threads 8
参数说明:
- -query: 输入文件路径及文件名
- -out:输出文件路径及文件名
- -db:格式化了的数据库路径及数据库名
- -outfmt:输出文件格式,总共有12种格式,6是tabular格式对应BLAST的m8格式
- -evalue:设置输出结果的e-value值
- -num_descriptions:tabular格式输出结果的条数
- -num_threads:线程数
四:输出文件解读
重点是-outfmt 6,也就是之前版本的m 8格式
结果中从左到右每一列的意义分别是:
- [00] Query id
- [01] Subject id
- [02] % identity
- [03] alignment length
- [04] mismatches
- [05] gap openings
- [06] q. start
- [07] q. end
- [08] s. start
- [09] s. end
- [10] e-value
- [11] bit score