全球最大的三阴性乳腺癌队列多组学数据说了什么
首发于生信技能树微信公众号:https://mp.weixin.qq.com/s/wWFYLBIfexaP1PuhqgcA9w
最近朋友圈被复旦大学邵志敏团队的三阴性乳腺癌队列研究刷屏了,比较中规中矩的报道大概是:

比较媒体风格的就是:

当然很容易勾起来我的兴趣,但是我看了几乎所有的报道,发现大家其实都没有说清楚这个研究到底有啥用。比如下面的描述:
研究团队针对465例三阴性乳腺癌标本展开研究,绘制出全球最大的三阴性乳腺癌队列多组学图谱。通过对庞大基因数据的分析,研究团队证实三阴性乳腺癌的确不是传统认识中的单一类型——三阴性乳腺癌不仅拥有自己的“家族”,家族中有不同的亚型,且不同亚型之间可能存在生存差异、对不同治疗方案敏感性不同。
实际上,这个是众所周知的,TNBC当然有不同的亚型,仅仅是从转录组视频就有十多篇分子分型的研究啦。
至于那些: 这项研究将为后续开展针对国人三阴性乳腺癌的药物研发、临床试验提供数据和证据的支持;后期我们也将针对这些特征进行更深层次的研究。
无非就是发表科研论文的官话罢了。
那我们也来看看这篇文章到底做了啥吧。邵志敏科研团队
其实他们团队连续发表了两篇文章:
首先是文章:Multi-omics profiling reveals distinct microenvironment characterization and suggests immune escape mechanisms of triple-negative breast cancer 里面提到了数据:
The sequencing data is also available in GSE118527 (OncoScan), GSE76250 (HTA 2.0) and SRP157974 (WES and RNAseq)
然后是文章:Genomic and Transcriptomic Landscape of Triple-Negative Breast Cancers: Subtypes and Treatment Strategies 里面提到了数据:
Microarray data and sequence data have also been deposited in the NCBI Gene Expression Omnibus (OncoScan array; GEO: GSE118527) and Sequence Read Archive (WES and RNA-seq; SRA: SRP157974).
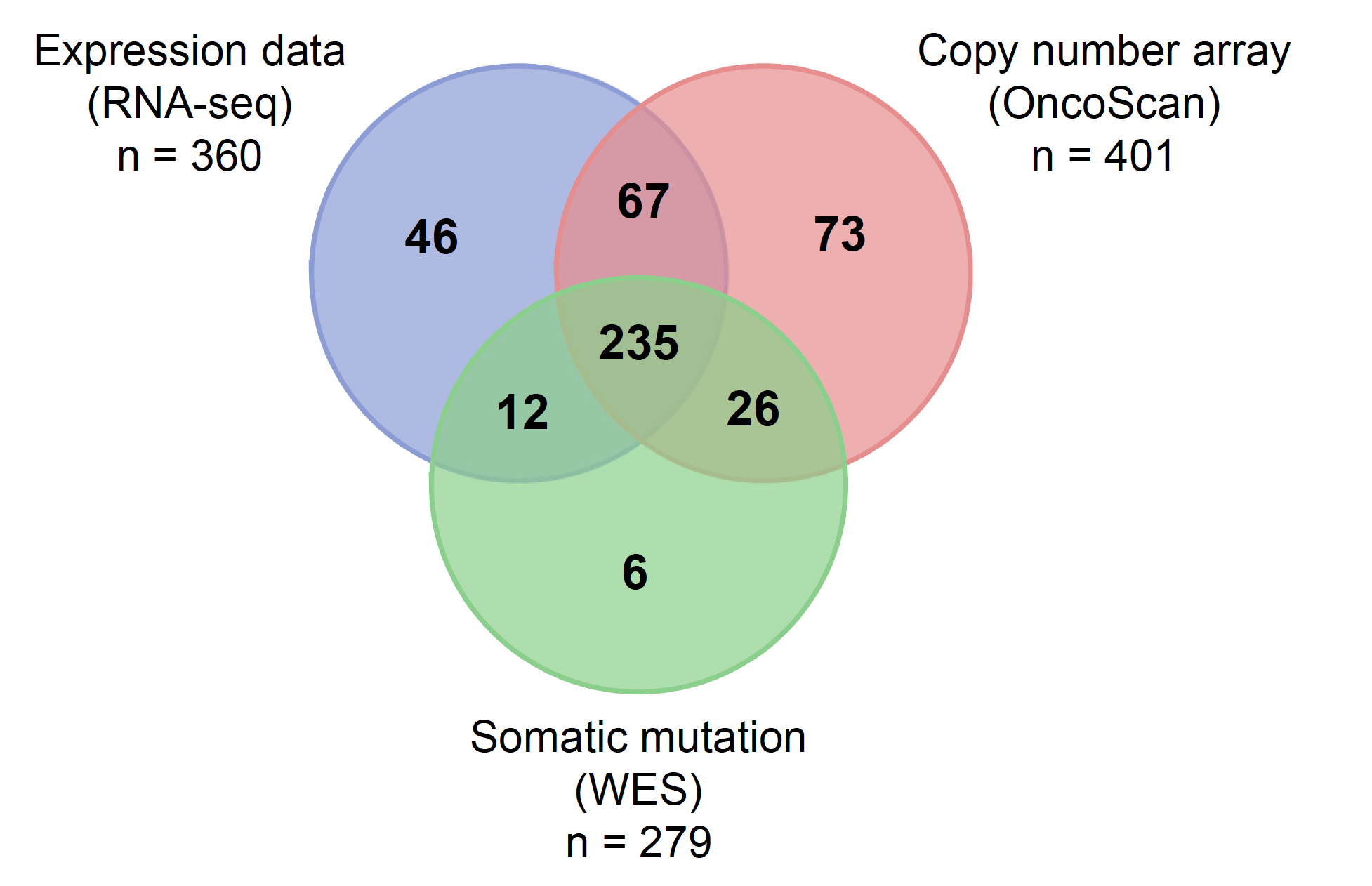
纳入的病人是:
A final cohort of 465 patients were available for analysis
- 279 had whole exome sequencing (WES) data on primary tumor tissue and paired blood samples
- 401 had copy-number alteration (CNA) data
- 360 had RNA sequencing data on primary tumor tissue.
中间还有个小插曲,因为这篇文章是先online,并没有正式见刊,所以当时我看到这个ID实际上去数据库是找不到的,发邮件给第一作者后,她回复很迅速,好在现在是可以下载原始数据了。
其中第一篇文章用到的公共数据也是他们团队自己做的,GSE76250 (HTA 2.0) 这个芯片表达数据,做的是:165 TNBC samples and 33 paired normal breast tissues, 他们团队分析的很透彻,连续发表了两篇文章,说明他们在这个领域深耕很久了- Liu YR, Jiang YZ, Xu XE, Hu X et al. Comprehensive Transcriptome Profiling Reveals Multigene Signatures in Triple-Negative Breast Cancer. Clin Cancer Res 2016 Apr 1;22(7):1653-62. PMID: 26813360 主要是分析生存资料。
- Jiang YZ, Liu YR, Xu XE, Jin X et al. Transcriptome Analysis of Triple-Negative Breast Cancer Reveals an Integrated mRNA-lncRNA Signature with Predictive and Prognostic Value. Cancer Res 2016 Apr 15;76(8):2105-14. PMID: 26921339 首先把配对的33个TNBC病人数据找差异,然后去137个不配对的样本里面验证,最后再招募82个病人探索疗效。
TNBC分型历史
看到很多媒体宣传最难治的乳腺癌有望获得分类治疗,但是其实TNBC分子分型的研究不少了,这个中国人TNBC队列既不是第一个,也不会是最后一个。
首先是2011的meta分析,把TNBC分成6类:- Basal-like 1 (BL1), basal-like 2 (BL2), immunomodulatory (IM), mesenchymal (M), mesenchymal stem-like (MSL) and luminal androgen receptor (LAR)
- 然后同样的作者2016年在plos one 发文重新修订了 之前的分类,变成4类: (TNBCtype-4) tumor-specific subtypes (BL1, BL2, M and LAR)
- 发表在Clin Cancer Res 2015 ,贝勒医学院研究小组的 Burstein 等人对自己的数据,198个TNBC病人芯片表达矩阵,使用80个核心基因进行分组,得到4个TNBC的亚型。
- 发表在 Breast Cancer Research (2015) :Gene-expression molecular subtyping of triple-negative breast cancer tumours: importance of immune response,数据在 GSE58812, 法国研究团队的等人使用 适应性的Fuzzy-clustering 把107个TNBC**患者分成3类。
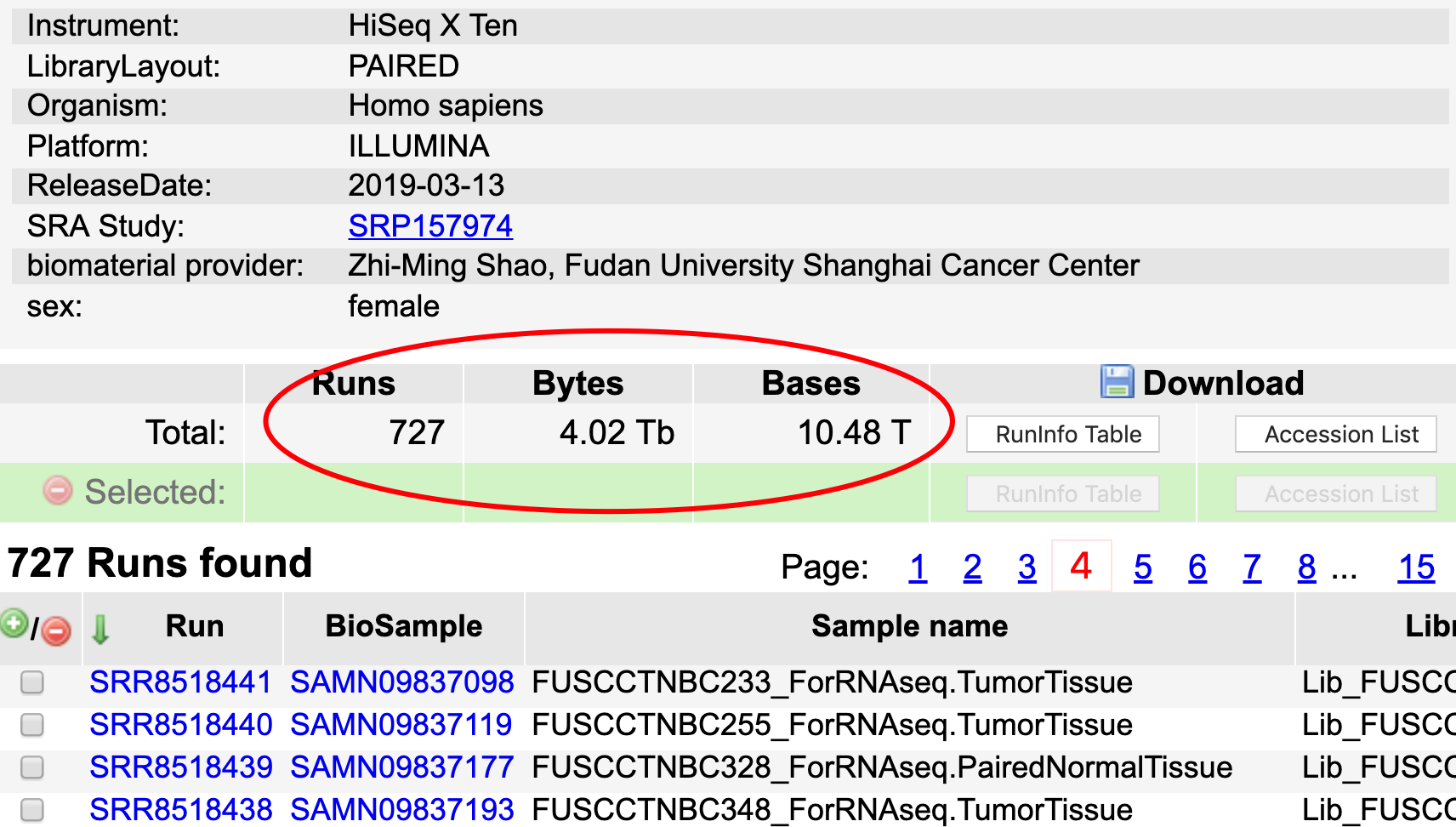
肿瘤外显子和转录组测序数据
数据都在 https://www.ncbi.nlm.nih.gov//sra/?term=SRP157974 ,数据量很可观,没有大服务器的就不用尝试重复这个分析了。
我已经把数据下载完了。突变全景图
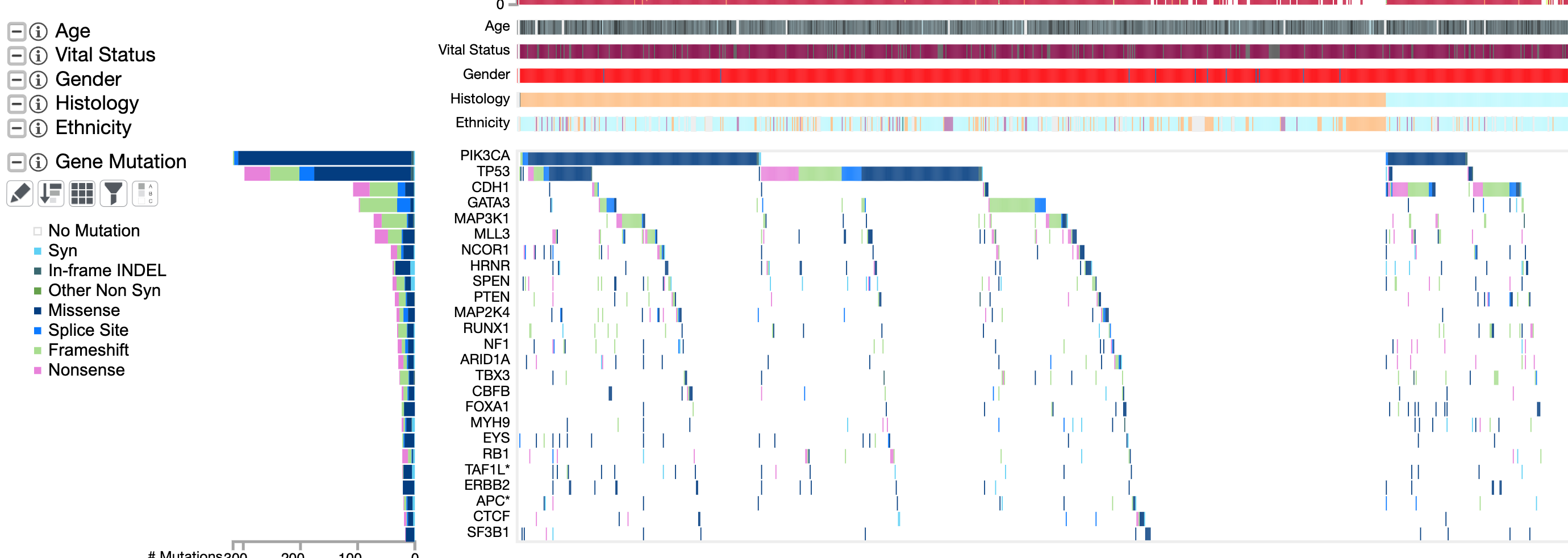
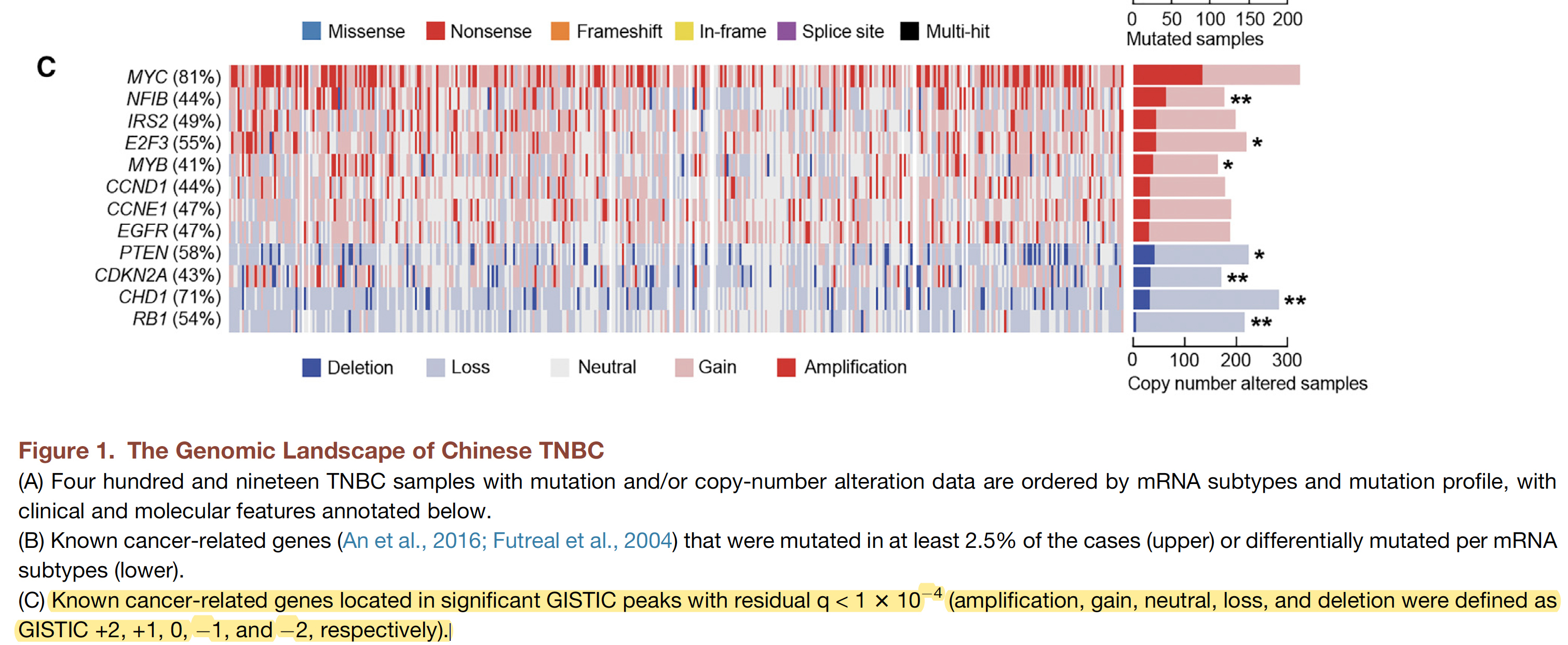
一般稍微有点样本量的肿瘤外显子队列,展示突变全景图已经成为了惯例,如下:
上面的TNBC已经按照表达量分成了4组。
这个时候,我们可能会与TCGA队列比较,见:http://firebrowse.org/iCoMut/?cohort=BRCA
当然了,TCGA数据库里面关于TNBC的样本也就一百来个,还有一个比较大的乳腺癌队列是METABRIC,也不多,而且并没有全部的基因突变信息,只有173个基因。
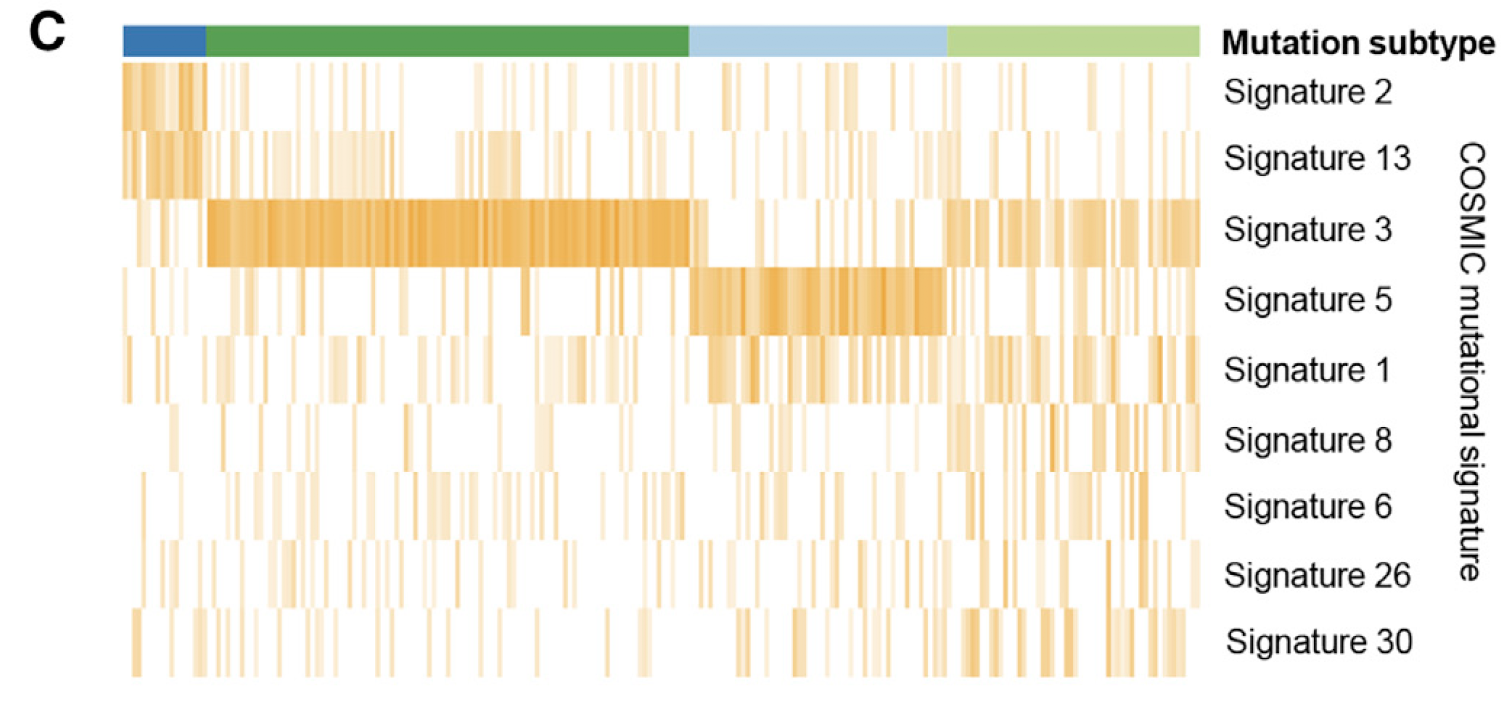
突变可以分解为了 COSMIC 数据库记录的 30个 signatures,每个样本的不同signatures比例如下:
比较有趣的是表达量分组,正好跟这个突变的signatures分组吻合。
当然,同步也可以看CNV信息,如下:
表达量分析
前面我们介绍了TNBC分子分型的历史,那么当然就作者会把自己的分组结果跟最经典的进行比较:
可以看到,如果根据作者的队列转录组数据进行分组,2011的meta分析把TNBC分成6类被这次的4类融合了。
说明了啥呢,2011的meta分析后面的分子分型研究也是跟他比较啊,我感觉这完全就是常规分析啊!
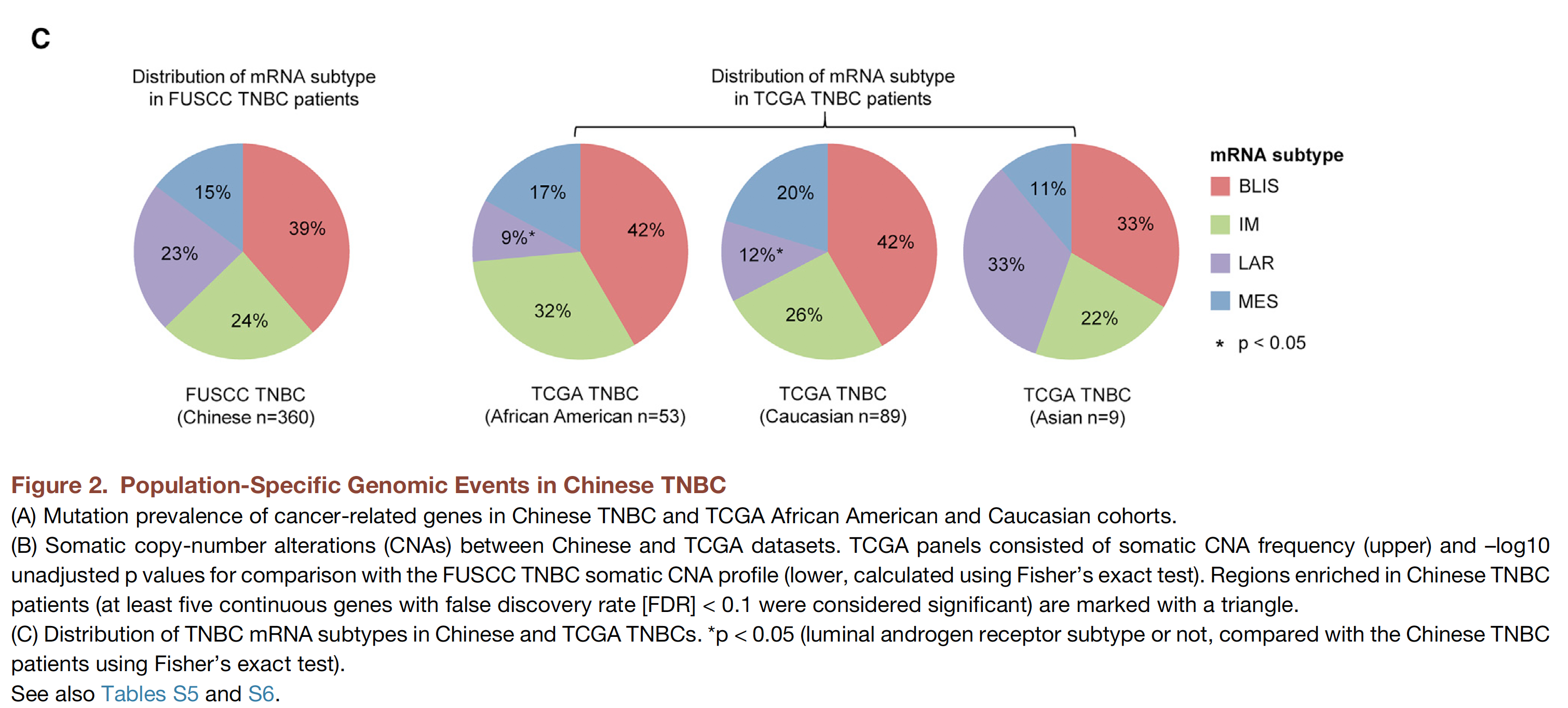
同样的,也是可以跟TCGA进行比较咯:
可以看到,TNBC的不同亚型,在不同人群的分布是有差异的
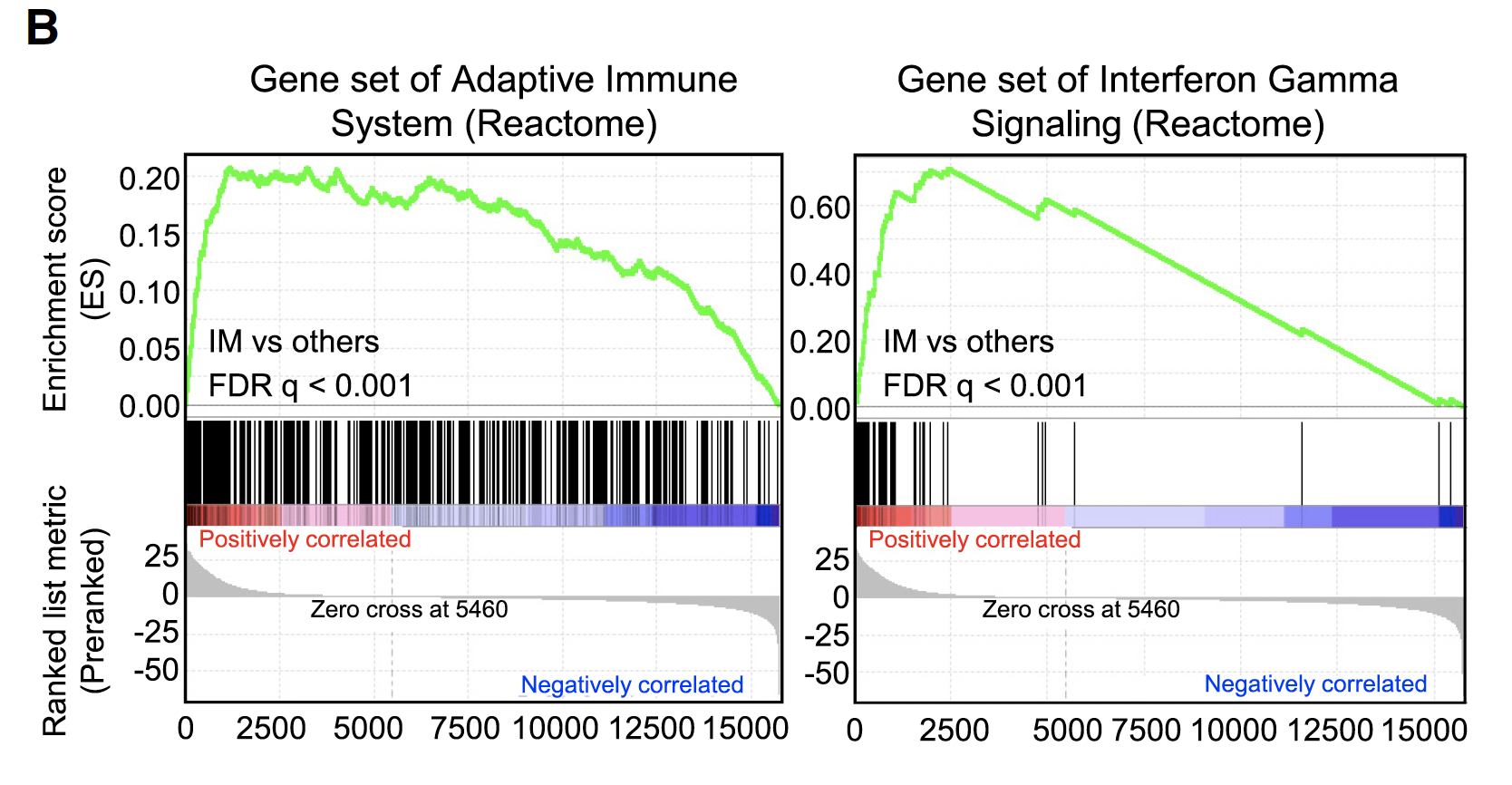
然后,研究者最关心的是免疫浸润亚型,所以可以拿这个组跟所有其他病人进行差异分析,后面就顺理成章的GSEA分析,然后富集的通路是免疫相关的,这个应该是理所当然吧!
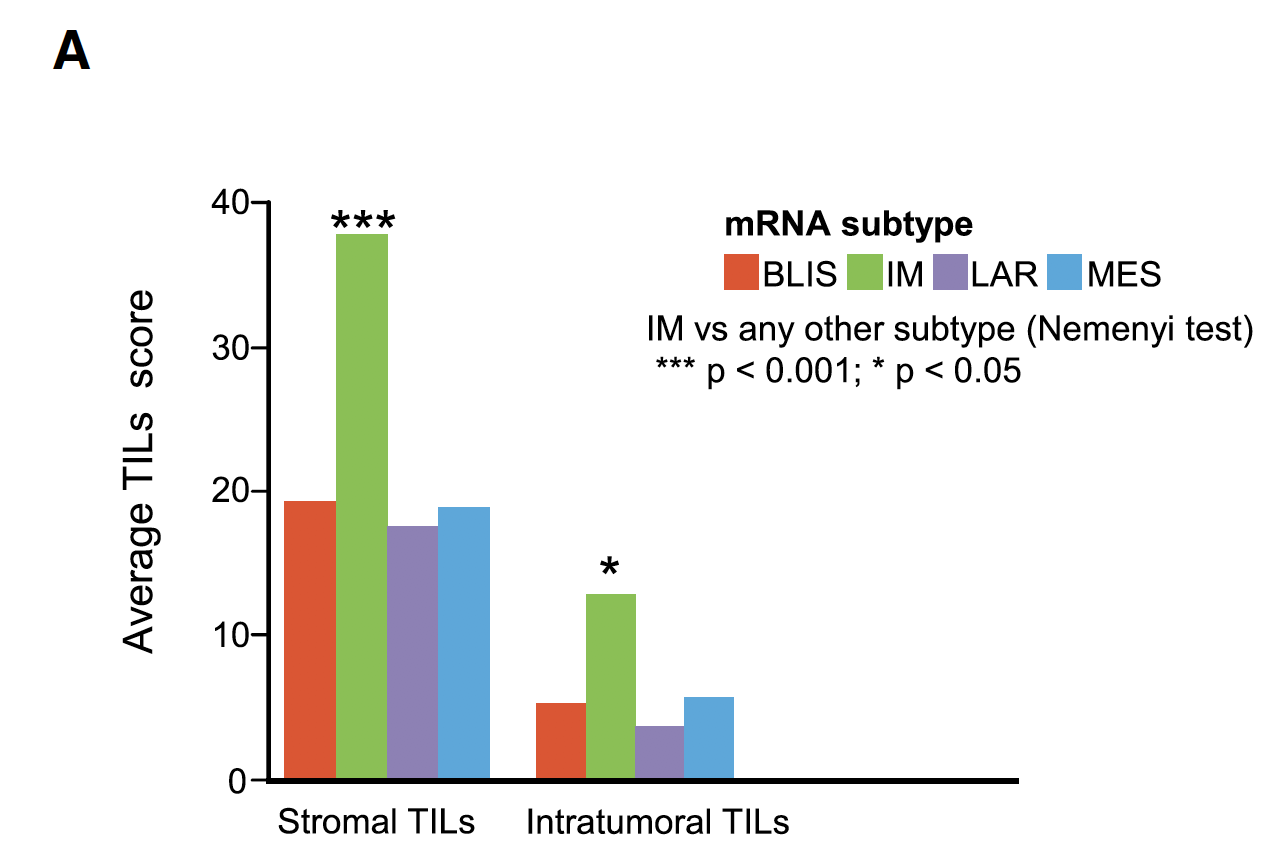
然后同样的,可以加上一个热点分析,就是免疫浸润细胞比例,也是有成熟的算法,同样也是描述一下不同分子亚型的区别,我没有看到最后的意义。
后面还有一些联合分析,突变后的基因的表达量如何。我的疑问
这样的NGS队列基本上来说,只要有病人资源和经费,就可以做,看起来呢,好像是得出了很多结论,实际上仔细看,又不知道到底其具体的贡献是什么?
亲爱的读者,你们觉得呢?