写在前面
昨天发了一个差点捅了马蜂窝的推文:费九牛二虎之力也无法重现的GSEA图 ,当时的确花了两三个小时也没能成功重复文章的GSEA图,觉得很有趣,就跟粉丝分享了,纯粹是交流数据处理技巧。
后来跟文章作者充分交流了,也理解到了他们研究的亮点,并不是在这个简单的芯片数据上面,而且不同的数据处理方法的选择,本身就会有不同的结果,但并不会影响他们文章的重大发现。
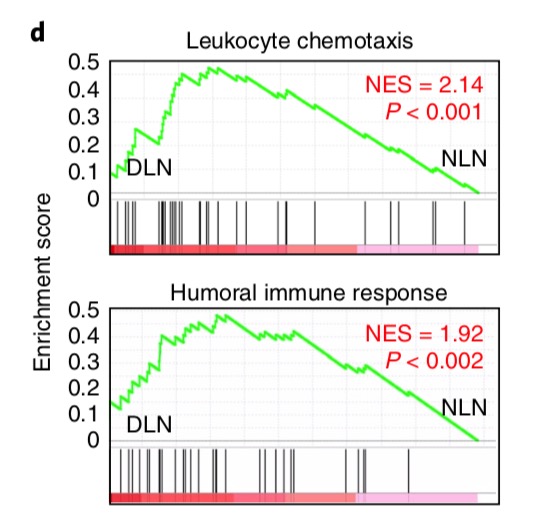
那我们现在就继续讨论一下,文中的GSEA的显著性结果图(下图)到底该如何来重现。
基因集大小的问题
请大家再注意看图中的基因集,应该是50个基因左右的数量,我昨天主要是就纠结在这里,因为我在不同版本的GO数据库资源,都找不到50个基因左右的数量的这两个通路,大家也可以自行去数据库搜索了解这两个GO基因集:
# GO:0002690 Positive regulation of leukocyte chemotaxis.
# http://amigo.geneontology.org/amigo/term/GO:0030595
# Accession GO:0030595
# Name leukocyte chemotaxis
# Ontology biological_process
# Synonyms immune cell chemotaxis, leucocyte chemotaxis
# http://amigo.geneontology.org/amigo/term/GO:0006959
# Accession GO:0006959
# Name humoral immune response
# Ontology biological_process
跟作者沟通
不懂就要问,所以我直接和文章的第一作者顾博士聊了一下这个数据问题,了解到了他们完成研究课题的艰辛和付出。只是术业有专攻,他们专注湿实验以及科研想法,在数据分析方面略微薄弱,也忽略了其中的细节描述。然后我们就干脆直接联系了完成芯片服务的公司,拿到了全部数据分析资料。
其中一张图文并茂的指示引起了我的注意:
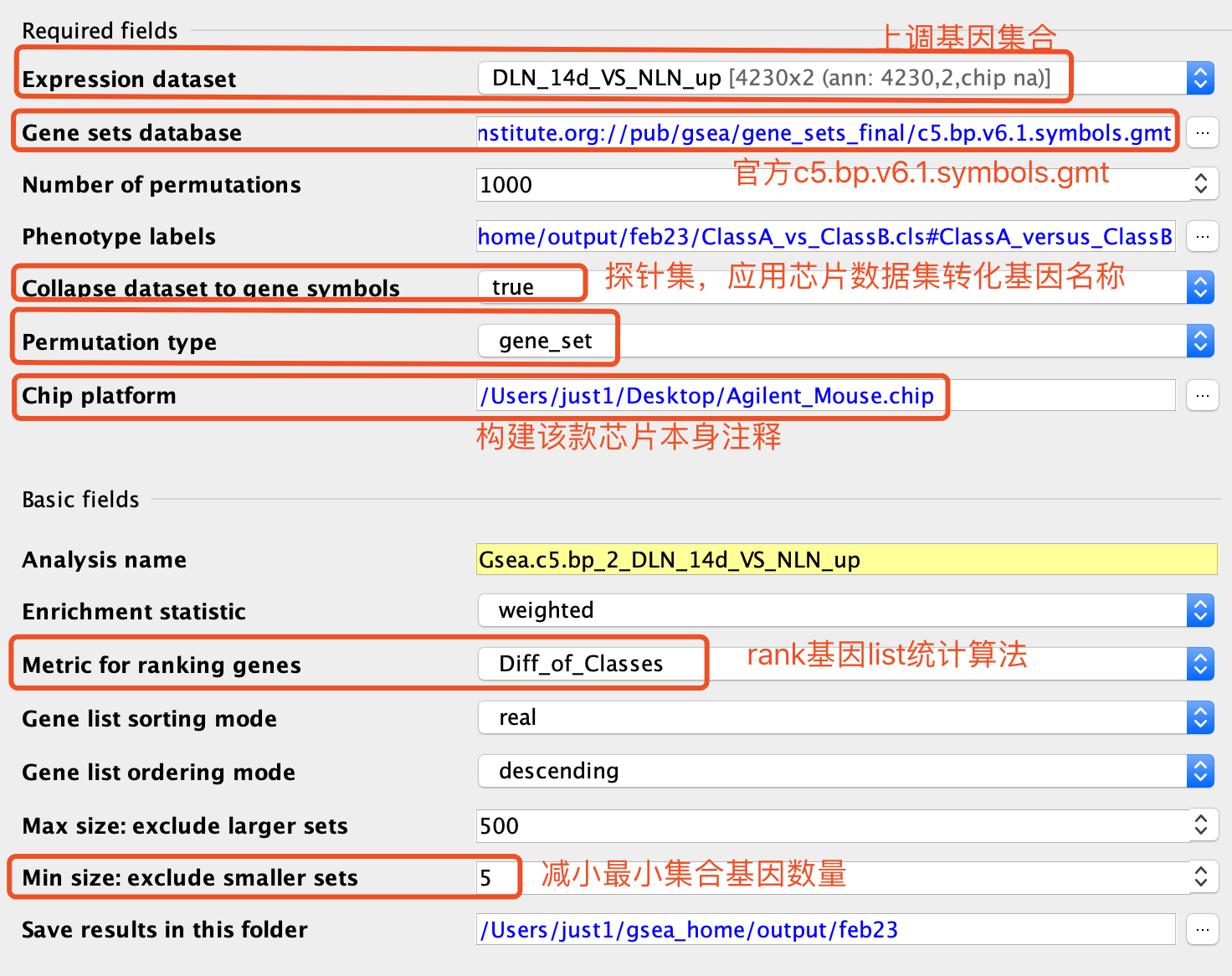
图中强调了是上调基因集合,作为背景基因,然后我刹那间相通了为什么他们图中是50个基因左右的数量的这两个通路,因为背景基因由两万多个被挑选为上调基因的4000多个,所以通路基因集必然也会缩水很多。
再次回顾他们的原始图:
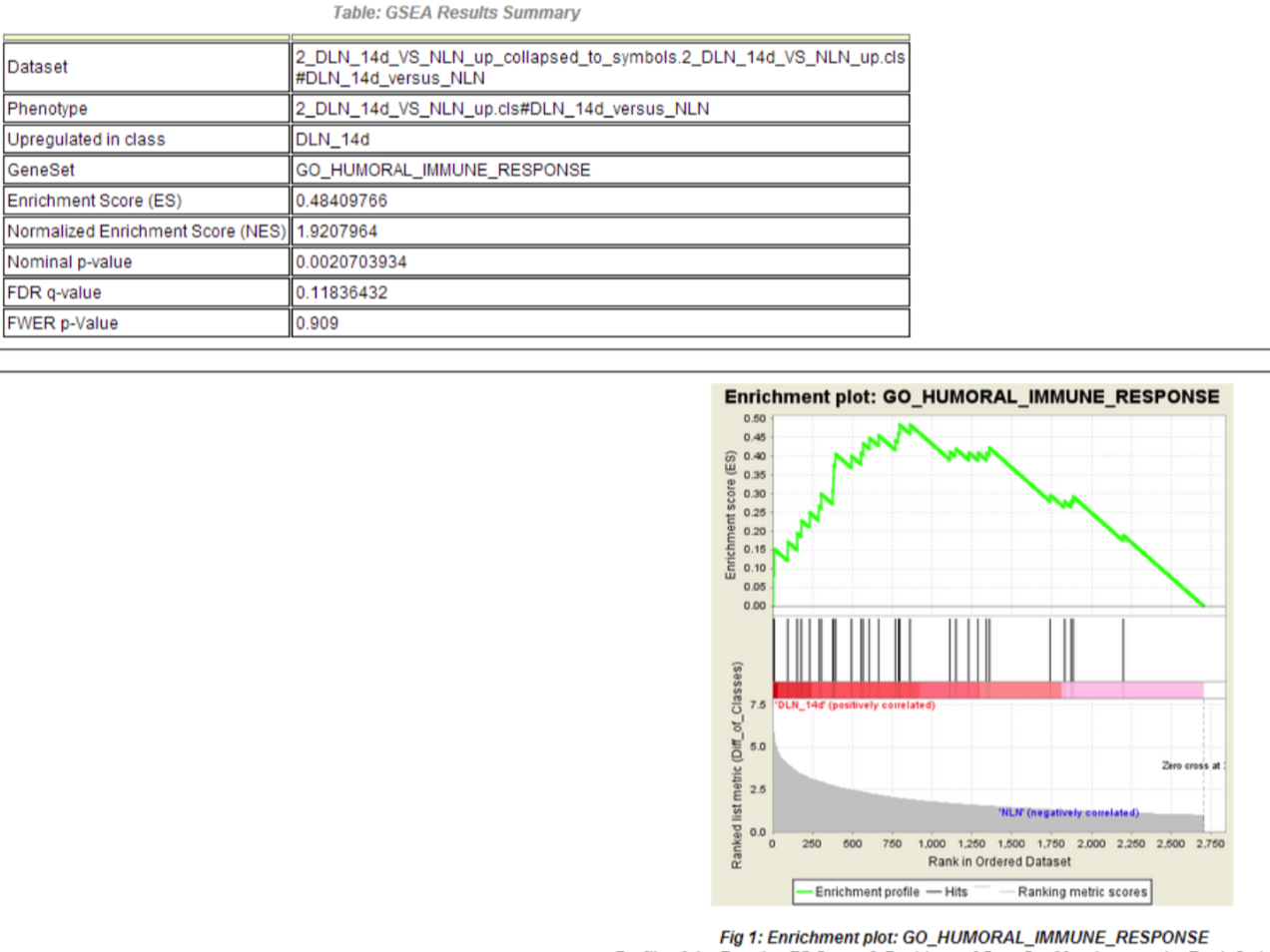
的确,p值和NES值都对,就是基因只有2700左右,很明显,他们是选择了上调探针的4000多个,但是只有其中的2700左右个探针是可以注释到基因的(关于这个探针注释到基因,是另外一个坑,跟本文无关)
既然知道了他们的策略,那么我就很容易写代码重复这个分析:
rm(list = ls()) ## 魔幻操作,一键清空~
options(stringsAsFactors = F)
library(GEOquery)
library(ggplot2)
library(clusterProfiler)
library(GSEABase)
load(file = 'step1-output.Rdata')
colnames(dat)
dat[1:4,1:4]
m=dat[,2]+dat[,1]/2
logFC1=log2(dat[,2]/dat[,1]);fivenum(logFC1)
logFC2=dat[,2]-dat[,1];fivenum(logFC2)
fivenum(logFC2)
## 这里我们也挑选基因
table(logFC2>0.7)
logFC2=logFC2[logFC2>0.7]
geneList=scale(logFC2)
names(geneList)=rownames(geneList)
geneList=sort(geneList,decreasing = T)
head(geneList)
# 这里需要注意的是 要 以 entrez ID来进行后续GSEA分析
# 如果是symbol,需要使用其它包,比如 GSEABase
library(org.Mm.eg.db)
s2g=select(org.Mm.eg.db,names(geneList),
'ENTREZID','SYMBOL')
head(s2g)
s2g=s2g[!is.na(s2g$ENTREZID),]
s2g=s2g[s2g$SYMBOL %in% names(geneList),]
geneList=geneList[s2g$SYMBOL]
names(geneList)=s2g$ENTREZID
head(geneList)
tail(geneList)
## 这里比较耗时:
if(F){
go_bp_gsea <- gseGO(geneList = geneList,
OrgDb='org.Mm.eg.db',
ont = 'BP',
nPerm = 1000,
minGSSize = 10,
pvalueCutoff = 0.9,
verbose = FALSE)
gseaplot(go_bp_gsea, geneSetID = rownames(go_bp_gsea[1,]))
gseaplot(go_bp_gsea, geneSetID = "GO:0006959")
gseaplot(go_bp_gsea, geneSetID = "GO:0030595")
tmp=go_bp_gsea@result
table(tmp$pvalue<0.01)
}
看其中一个结果,可以看到几乎再现了作者的结果,现在的差异就来自于我们选取的基因数量不一样,但都是根据logFC值来挑选了上调基因后做的GSEA分析,我们需要讨论这样的合理性。
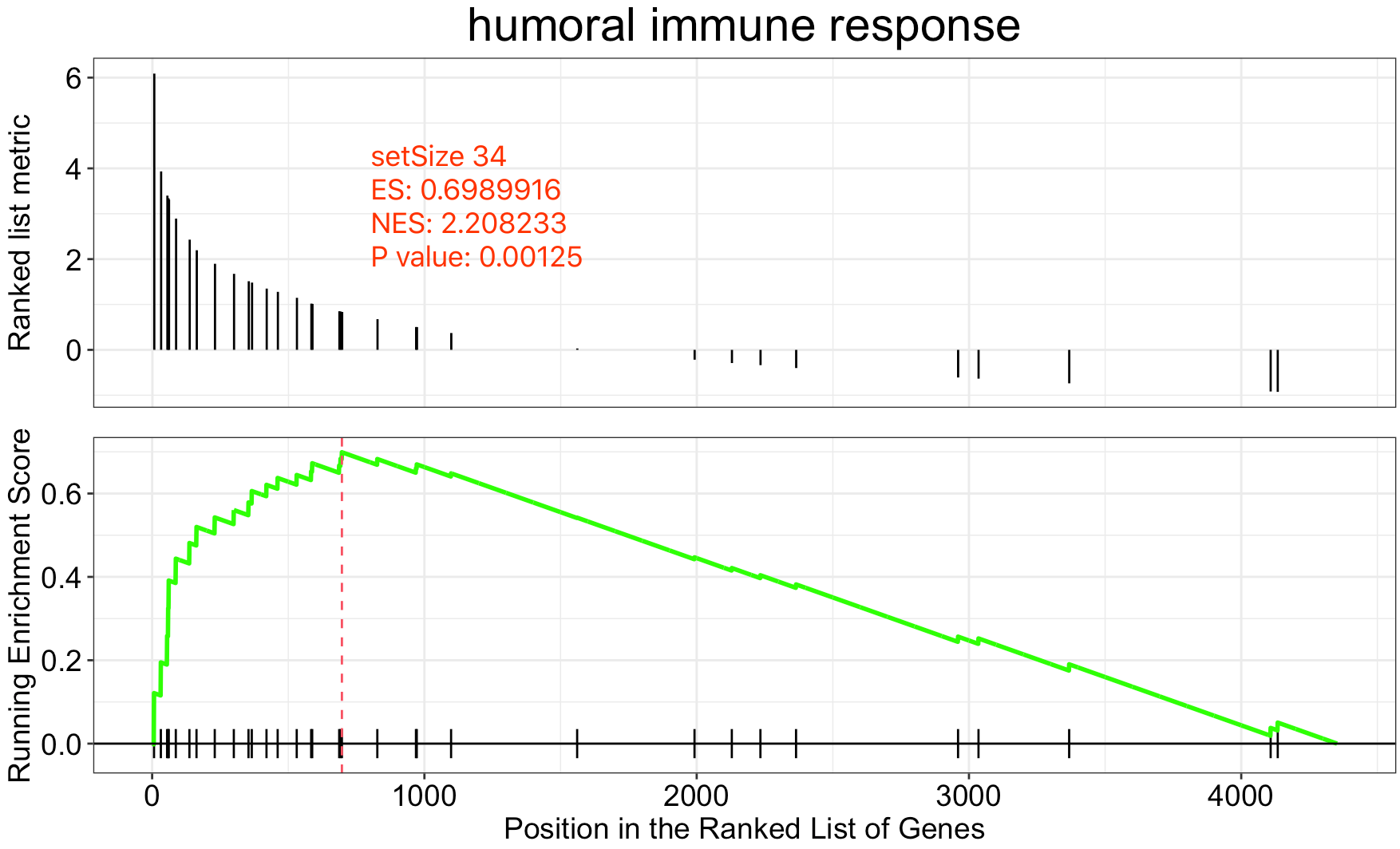
挑选上调基因集是否合理呢
虽然我这边重复出来了作者的GSEA显著结果,但是并不意味着万事大吉,我们仍然要讨论一下研究者所委托的公司使用这样的策略来做GSEA分析是否合理。
首先,在GSEA官网文档,很清楚的描述:
The GSEA algorithm does not filter the expression dataset and does not benefit from your filtering of the expression dataset. During the analysis, genes that are poorly expressed or that have low variance across the dataset populate the middle of the ranked gene list and the use of a weighted statistic ensures that they do not contribute to a positive enrichment score. By removing such genes from your dataset, you may actually reduce the power of the statistic.
也就是说,不建议自行过滤基因,更别说是挑选上调表达基因了。
这个公司的策略是挑选上调基因,再去做gsea看某些通路是否富集,的确是合理性不足!
但是顾博士给我一个链接:
Although GSEA does not require that you preprocess the expression dataset, it can be used effectively on preprocessed datasets. For example, Monti et al used a filtered dataset to further analyze genes consistently expressed across two datasets
参考文献是:“Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response” (http://www.bloodjournal.org/cgi/content/full/bloodjournal;105/5/1851).
里面提到了作者收集整理了相关领域的4个研究的281个基因集,但是他们的GSEA方法,就是挑选基因后的GSEA,Enrichment was assessed by:
- (1) ranking the 2118 genes in the top 5% pool with respect to the phenotype “cluster X versus not cluster X”;
- (2) locating the represented members of a given gene set within the ranked 2118 genes;
一些讨论
关于公司这个GSEA分析策略的统计学合理性,在我们生信技能树VIP群有着激烈的讨论:
药企东哥发言:
很多研究领域已经有了确定的发现,这些背景知识可以作为positive control用于判断以及合理的调整分析策略。跟研究目的也有关吧。不一定非得为了“totally unbiased result”闭着眼跑“标准流程”,毕竟大多数生物信息方法都是不完美的。分析结果可解释很重要
其他人发言:
略