组会有同学分享了曹雪涛课题组的一篇Nature Medicine文章,其生物学机理探索非常详细,但是我注意到他们使用芯片技术仅仅是测了才4个样本,就区分成两种细胞,两种处理,也就是说根本没有生物学重复,我相信他们课题组应该是不差钱的,那很可能是他们并没有意识到NGS技术的重要性。
不过,我这里不是要批评谁,我就是感兴趣这个文章是如何使用这个数据集的,后面也有几个小问题,感兴趣的粉丝可以来信跟我交流哈。
文章介绍
转移是肿瘤致死的原因之一。在肿瘤转移的过程中,原发部位肿瘤细胞可以刺激骨髓来源的细胞(称为被肿瘤驯化的细胞),促使并招募其进入远端拟转移部位,形成转移前微环境,为肿瘤细胞转移至该部位提供合适“土壤”。因此识别被肿瘤驯化的细胞,明确其功能是揭露肿瘤发生发展的关键步骤。
2019年1月15日凌晨,曹雪涛院士团队在Nature Medicine杂志发表了题为Tumor-educated B cells selectively promote breast cancer lymph node metastasis by HSPA4-targeting IgG的最新研究成果,详细揭示了B细胞能够通过分泌靶向肿瘤抗原HSPA4(Heat shock protein 4)的病理性抗体促进乳腺癌淋巴结转移。
本研究阐明了B细胞及抗体介导的体液免疫在淋巴结转移前微环境形成及肿瘤淋巴结转移中的重要功能。首次发现除调节性B细胞介导的负向免疫调控功能外,B细胞能够通过分泌靶向肿瘤抗原的病理性抗体直接促进肿瘤转移,同时寻找到糖基化的肿瘤膜抗原在病理性抗体的产生及促转移中的重要功能,为深入认识体B细胞介导的体液免疫功能及肿瘤转移前微环境的形成提供了新的视角,为肿瘤治疗尤其是肿瘤转移的预测和防治提供了新的靶点。
芯片实验设计及文章数据处理结果
值得注意的是研究者仅仅是测了才4个样本,就区分成两种细胞,两种处理,也就是说根本没有生物学重复。

从文章里面拿到了芯片信息,再看看其热图的描述:
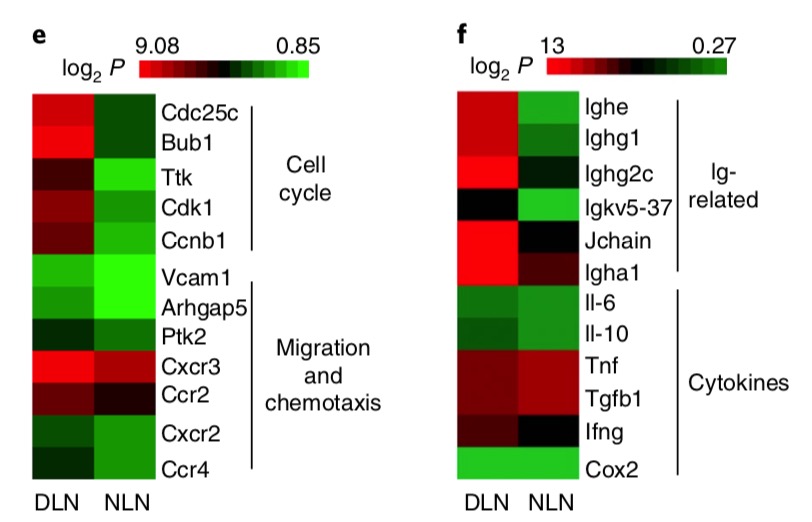
# lymph nodes from normal mice (NLNs)
# draining lymph nodes (DLNs)
# cell cycle genes (such as Cdc25c, Bub1, Ttk, and Cdk1) and
# migration-related genes (such as Vcam1, Arhgap5, Cxcr3 and Ccr2)
# were upregulated in DLN B cells compared with NLN B cells
# Heat map of genes expressed in purified B cells
# (CD19+) from NLNs and DLNs as determined by gene chip array
# (n = 1 for gene chip analysis of d−f)
是B细胞的数据的指定的两个基因集,奇怪的是 作者的图例是 log2P值,很诡异!!!
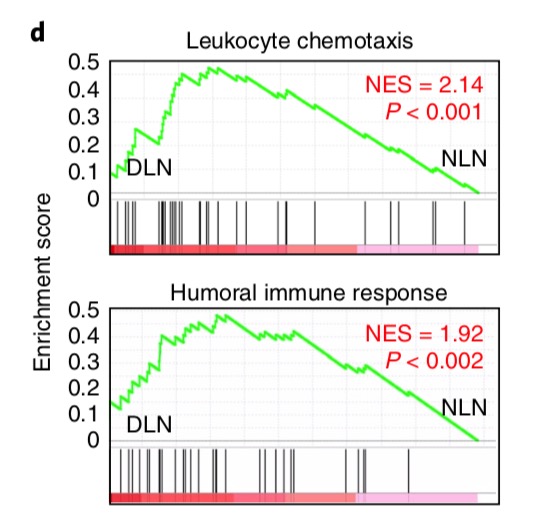
再看看GSEA结果:
# d, Gene set enrichment analysis (GSEA) analysis for gene signatures of
# leukocyte chemotaxis and humoral immune response
# in purified stromal cells (CD45−) from DLN compared with NLN control.
# NES, normalized enrichment score.
这里是stromal细胞的数据了。
作者绘制的是两个GO基因集:
# GO:0002690 Positive regulation of leukocyte chemotaxis.
# http://amigo.geneontology.org/amigo/term/GO:0030595
# Accession GO:0030595
# Name leukocyte chemotaxis
# Ontology biological_process
# Synonyms immune cell chemotaxis, leucocyte chemotaxis
# http://amigo.geneontology.org/amigo/term/GO:0006959
# Accession GO:0006959
# Name humoral immune response
# Ontology biological_process
后面我们要重复这种图就需要有这样的背景知识,注意看图中的基因集,应该是50个基因左右的数量。
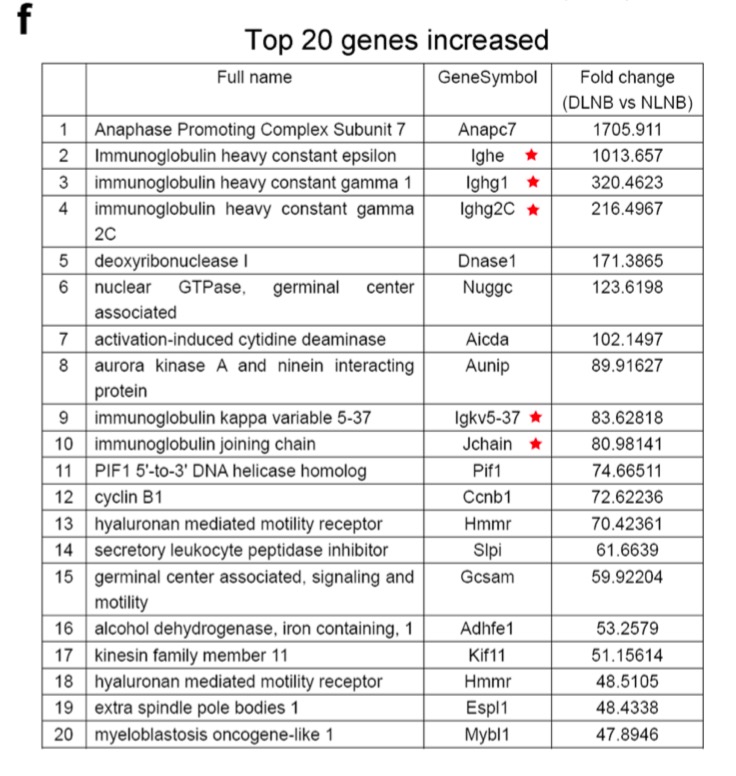
最后,作者列了一下排名前20的差异基因,如下:‘
文章结果复现
表达矩阵的下载很容易:
rm(list = ls()) ## 魔幻操作,一键清空~
options(stringsAsFactors = F)
library(GEOquery)
# stromal cells from tumor-draining lymph nodes
gset <- getGEO('GSE113249', destdir=".",
AnnotGPL = F, ## 注释文件
getGPL = F) ## 平台文件
a=gset[[1]] #
dat=exprs(a) #a现在是一个对象,取a这个对象通过看说明书知道要用exprs这个函数
dim(dat)#看一下dat这个矩阵的维度
head(dat)
pd=pData(a) #通过查看说明书知道取对象a里的临床信息用pData
colnames(dat)=pd$title
dat1=dat
gset <- getGEO('GSE113250', destdir=".",
AnnotGPL = F, ## 注释文件
getGPL = F) ## 平台文件
a=gset[[1]] #
dat=exprs(a) #a现在是一个对象,取a这个对象通过看说明书知道要用exprs这个函数
dim(dat)#看一下dat这个矩阵的维度
head(dat)
pd=pData(a) #通过查看说明书知道取对象a里的临床信息用pData
colnames(dat)=pd$title
dat2=dat
dat=cbind(dat1,dat2)
dat[1:4,1:4]
boxplot(dat)
# GPL21163 Agilent-074809 SurePrint G3 Mouse GE v2 8x60K Microarray [Probe Name version]
if(F){
library(GEOquery)
#Download GPL file, put it in the current directory, and load it:
gpl <- getGEO('GPL21163', destdir=".")
colnames(Table(gpl))
head(Table(gpl)[,c(1,6)]) ## you need to check this , which column do you need
probe2gene=Table(gpl)[,c(1,6)]
save(probe2gene,file='probe2gene.Rdata')
}
load(file='probe2gene.Rdata')
ids=probe2gene
head(ids)
colnames(ids)=c('probe_id','symbol')
ids=ids[ids$symbol != '',]
ids=ids[ids$probe_id %in% rownames(dat),]
dat[1:4,1:4]
dat=dat[ids$probe_id,]
ids$median=apply(dat,1,median) #ids新建median这一列,列名为median,同时对dat这个矩阵按行操作,取每一行的中位数,将结果给到median这一列的每一行
ids=ids[order(ids$symbol,ids$median,decreasing = T),]#对ids$symbol按照ids$median中位数从大到小排列的顺序排序,将对应的行赋值为一个新的ids
ids=ids[!duplicated(ids$symbol),]#将symbol这一列取取出重复项,'!'为否,即取出不重复的项,去除重复的gene ,保留每个基因最大表达量结果s
dat=dat[ids$probe_id,] #新的ids取出probe_id这一列,将dat按照取出的这一列中的每一行组成一个新的dat
rownames(dat)=ids$symbol#把ids的symbol这一列中的每一行给dat作为dat的行名
dat[1:4,1:4] #保留每个基因ID第一次出现的信息
dim(dat)
save(dat,file = 'step1-output.Rdata')
load(file = 'step1-output.Rdata')
可以看到样本相关性与其分组是匹配的:
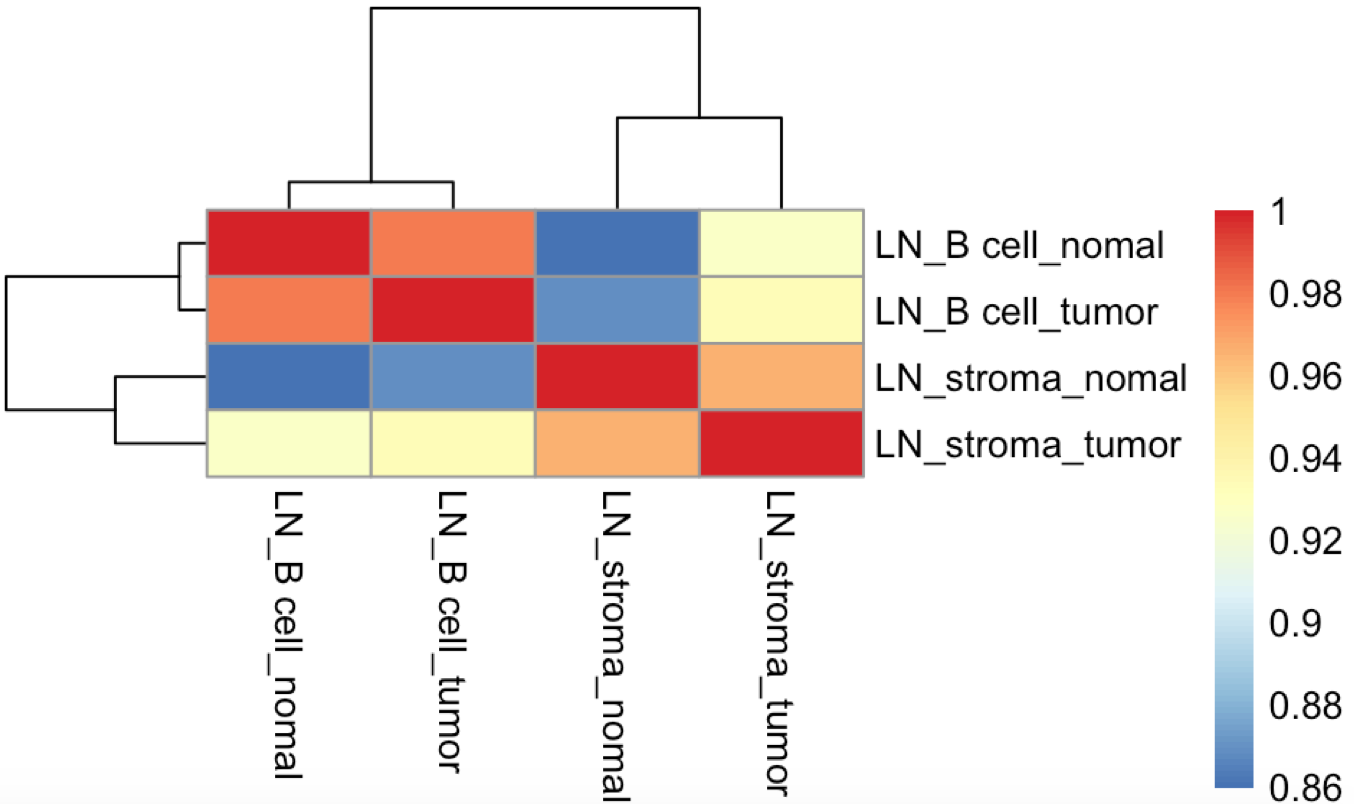
热图很容易重现,代码如下:
rm(list = ls()) ## 魔幻操作,一键清空~
options(stringsAsFactors = F)
library(GEOquery)
load(file = 'step1-output.Rdata')
colnames(dat)
dat[1:4,1:4]
library(pheatmap)
ccg=trimws(strsplit('Cdc25c, Bub1, Ttk, Cdk1',',')[[1]])
mrg=trimws(strsplit('Vcam1, Arhgap5, Cxcr3, Ccr2',',')[[1]])
mat=dat[c(ccg,mrg),3:4]
mat
pheatmap(mat)
pheatmap(dat[rownames(dat)[grepl('^Ig',rownames(dat))],])
主要是GSEA图,我首先拿出基因排序:
rm(list = ls()) ## 魔幻操作,一键清空~
options(stringsAsFactors = F)
library(GEOquery)
load(file = 'step1-output.Rdata')
colnames(dat)
dat[1:4,1:4]
rownames(dat)=toupper(rownames(dat))
m=dat[,2]+dat[,1]/2
logFC1=log2(dat[,2]/dat[,1]);fivenum(logFC1)
logFC2=dat[,2]-dat[,1];fivenum(logFC2)
plot(logFC1,m)
plot(logFC2,m)
plot(logFC1,logFC2)
# 这里的 logFC1 和 logFC2 差异很小
geneList=scale( logFC2 )r
# geneList=scale( dat[,2]/dat[,1] )
names(geneList)=rownames(dat)
geneList=sort(geneList,decreasing = T)
head(geneList)
所有的 gsea 分析都应该是基于 geneList这个对象,这个编程思想很重要。
然后创建基因集,我首先是要 gskb 这个R包
library(gskb)
#browseVignettes("gskb")
library(gskb)
data(mm_GO)
(g1=mm_GO$GO_BP_MM_HUMORAL_IMMUNE_RESPONSE)
(g2=mm_GO$GO_BP_MM_LEUKOCYTE_CHEMOTAXIS)
g1=g1[g1 %in% rownames(dat)]
g2=g2[g2 %in% rownames(dat)]
library(pheatmap)
pheatmap(dat[g1 ,1:2])
pheatmap(dat[g2 ,1:2])
然后进行GSEA分析:
library(ggplot2)
library(clusterProfiler)
library(GSEABase) # BiocManager::install('GSEABase')
# geneset <- read.gmt('h.all.v6.2.symbols.gmt')
# Then we know geneset is a simple data.frame
# first column is name of geneset, second column is gene symbol
geneset=data.frame(ont=c(rep('HUMORAL_IMMUNE_RESPONSE',length(g1)),
rep('LEUKOCYTE_CHEMOTAXIS',length(g2))),
gene=c(g1,g2))
egmt <- GSEA(geneList, TERM2GENE=geneset,
verbose=T)
head(egmt)
gseaplot(egmt,'HUMORAL_IMMUNE_RESPONSE')
gseaplot(egmt,'LEUKOCYTE_CHEMOTAXIS')
发现只有一个基因集是显著的,另外一个惨不忍睹,我就不发图了。
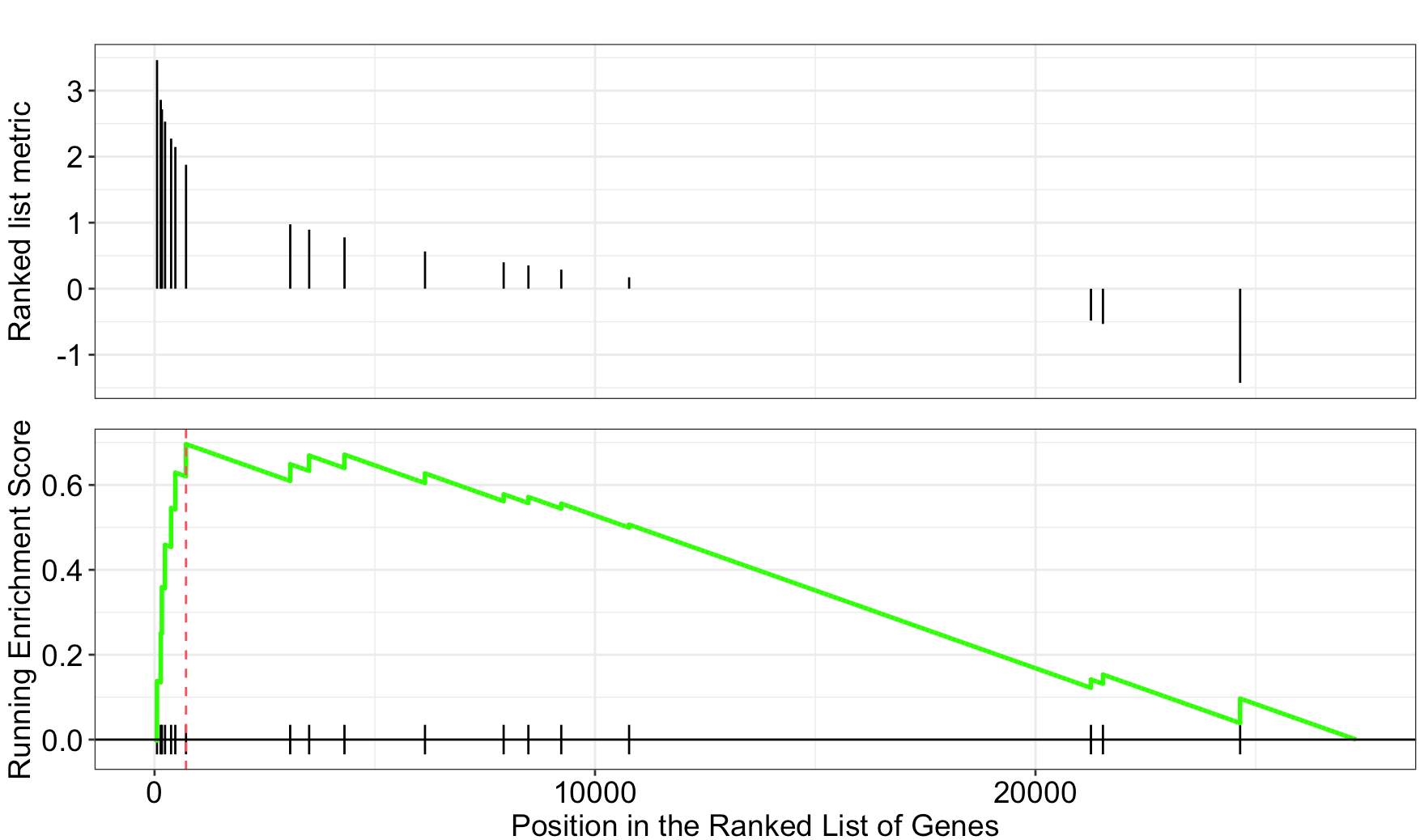
然后我尝试使用 GO.db 包的数据集。
library(GO.db)
ls("package:GO.db")
library(org.Hs.eg.db)
eg2go=toTable(org.Hs.egGO2ALLEGS)
head(eg2go)
tmp=eg2go[eg2go$go_id=='GO:0030595',]
columns(org.Hs.eg.db)
g11=select(org.Hs.eg.db,tmp$gene_id,'SYMBOL', 'ENTREZID')[,2]
g11=g11[g11 %in% rownames(dat)]
tmp=eg2go[eg2go$go_id=='GO:0006959',]
columns(org.Hs.eg.db)
g22=select(org.Hs.eg.db,tmp$gene_id,'SYMBOL', 'ENTREZID')[,2]
g22=g22[g22 %in% rownames(dat)]
library(pheatmap)
pheatmap(dat[g11 ,1:2])
pheatmap(dat[g22 ,1:2])
geneset=data.frame(ont=c(rep('HUMORAL_IMMUNE_RESPONSE',length(g11)),
rep('LEUKOCYTE_CHEMOTAXIS',length(g22))),
gene=c(g11,g22))
egmt <- GSEA(geneList, TERM2GENE=geneset,
verbose=T)
head(egmt)
gseaplot(egmt,'HUMORAL_IMMUNE_RESPONSE')
gseaplot(egmt,'LEUKOCYTE_CHEMOTAXIS')
发现也不是显著富集,最后我再使用Y叔的R包,这里面有个R代码技巧,很重要,值得大家花费几个小时慢慢理解。
rm(list = ls()) ## 魔幻操作,一键清空~
options(stringsAsFactors = F)
library(GEOquery)
load(file = 'step1-output.Rdata')
colnames(dat)
dat[1:4,1:4]
rownames(dat)=toupper(rownames(dat))
m=dat[,2]+dat[,1]/2
logFC1=log2(dat[,2]/dat[,1]);fivenum(logFC1)
logFC2=dat[,2]-dat[,1];fivenum(logFC2)
plot(logFC1,m)
plot(logFC2,m)
plot(logFC1,logFC2)
geneList=scale( logFC2 )
names(geneList)=rownames(dat)
geneList=sort(geneList,decreasing = T)
head(geneList)
s2g=select(org.Hs.eg.db,names(geneList),
'ENTREZID','SYMBOL')
head(s2g)
s2g=s2g[!is.na(s2g$ENTREZID),]
s2g=s2g[s2g$SYMBOL %in% names(geneList),]
geneList=geneList[s2g$SYMBOL]
names(geneList)=s2g$ENTREZID
head(geneList)
tail(geneList)
library(ggplot2)
library(clusterProfiler)
library(GSEABase)
## 这里比较耗时:
go_bp_gsea <- gseGO(geneList = geneList,
OrgDb='org.Hs.eg.db',
ont = 'BP',
nPerm = 1000,
minGSSize = 10,
pvalueCutoff = 0.9,
verbose = FALSE)
gseaplot(go_bp_gsea, geneSetID = rownames(go_bp_gsea[1,]))
gseaplot(go_bp_gsea, geneSetID = "GO:0006959")
gseaplot(go_bp_gsea, geneSetID = "GO:0030595")
得到的GSEA图,仍然是不显著。
思考
为什么我工具芯片表达量算的两种logFC的散点图出现了染色体一样的结构:
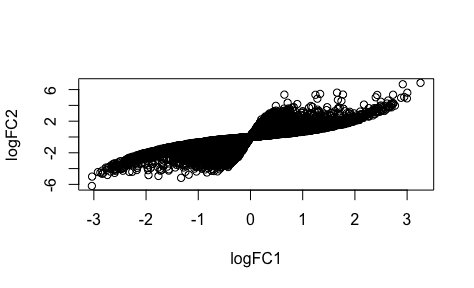
为什么使用不同数据库来源的同一个生物学功能基因集得到的结果可以千差万别。
为什么我最后也无法重复出来一个基因集的显著结果呢?是作者错了吗,还是我分析方法不对?
提示
我默认对同一个基因对应的多个探针, 选取了表达量最大的探针,但是我在检查作者的top20基因的时候发现,特别诡异的是我看到的那些基因表达量并没有差异,所以我怀疑是 选取了表达量最大的探针来进行过滤这个策略恐怕是有问题的。
比如 Anapc7 这个基因是有3个探针的,但是:
#
# A_52_P384134 Anapc7
# A_55_P2112330 Anapc7
# A_55_P2731736 Anapc7
dat['A_52_P384134',]
dat['A_55_P2112330',]
dat['A_55_P2731736',]
我恰好挑的探针是没有差异的,如下:
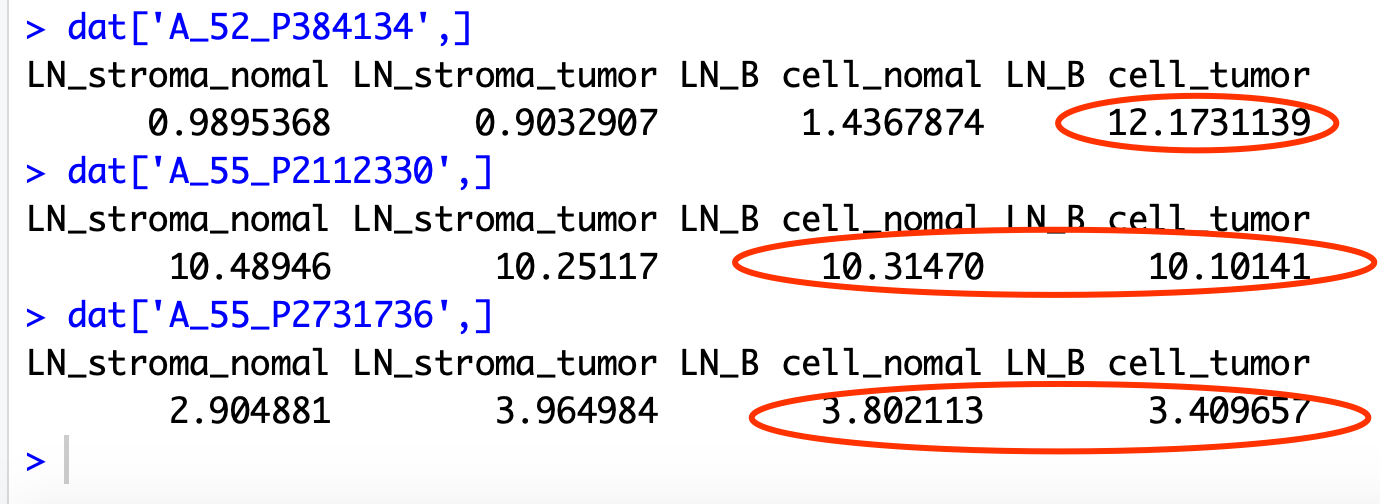
这样问题就来了,作者只测一个样本,这样的数值可靠吗?