于2017年发表在 Nature Communications 杂志,标题是:Phylogenetic analysis of metastatic progression in breast cancer using somatic mutations and copy number aberrations ,主要完成单位来自于比利时:
- Breast Cancer Translational Research Laboratory, Institut Jules Bordet, Université Libre de Bruxelles, Belgium
研究人员从10个尸检病人提取正常组织,原位癌组织以及多个转移瘤部位进行测序,总共是 51 samples (median coverage 40±18 × ) 。
每个病人都有着完善的信息记录,在其补充材料。
因为乳腺癌的随访和频繁取样较为困难,对乳腺癌转移(包括从原发肿瘤到癌细胞转移)的研究几乎不可能,因为这需要对所有患者的全部转移过程进行分析,所以从死于不同转移时期的乳腺癌患者尸体获得标本,是从整体水平明确该疾病转移特征的唯一手段。找somatic的mutation方法
全外显子测序找到的somatic的mutation只是初步结果,作者还用了两个验证手段。
We used whole-exome sequencing to index somatic mutations from 51 samples (median coverage 40±18 × ) followed by orthogonal validation using Sequenom MassARRAY to exclude false positive calls and targeted amplicon ultra-deep sequencing (median coverage 11,390±5,646 × ) to obtain accurate variant allele frequencies (VAFs).
正是因为如此完善的验证,可以把找到的SNV进行分类如下:SNVs的5级分类
分别如下:
- tier-0 SNVs as any SNV indexed in any sample including the matched germline reference by exome sequencing.
- Tier-1 SNVs are those that were further called somatic after comparison with the germline reference
- tier-2 SNVs are the subset of tier-1 which have been further validated and confirmed somatic by Sequenom MassARRAY.
- Tier-3 SNVs are the further subset that was confirmed present by targeted deep amplicon sequencing. >1500X coverage and >3% VAF
- the top-level of tier-4 SNVs using samples with >30% CCF, >1500X coverage and >3% VAF.
所以这里的分析重点是tier-3 SNVs拷贝数芯片
For the estimation of CNAs, a total of 64 samples were shipped for processing to the Affymetrix Research Services Laboratory. Normal and tumour DNA from FFPE samples were genotyped using the Affymetrix OncoScan FFPE Express 2.0 arrays.
使用的不是经典的ASCAT和ABSOLUT软件进行分析,而是GAP 方法,但是作者进行了比较。
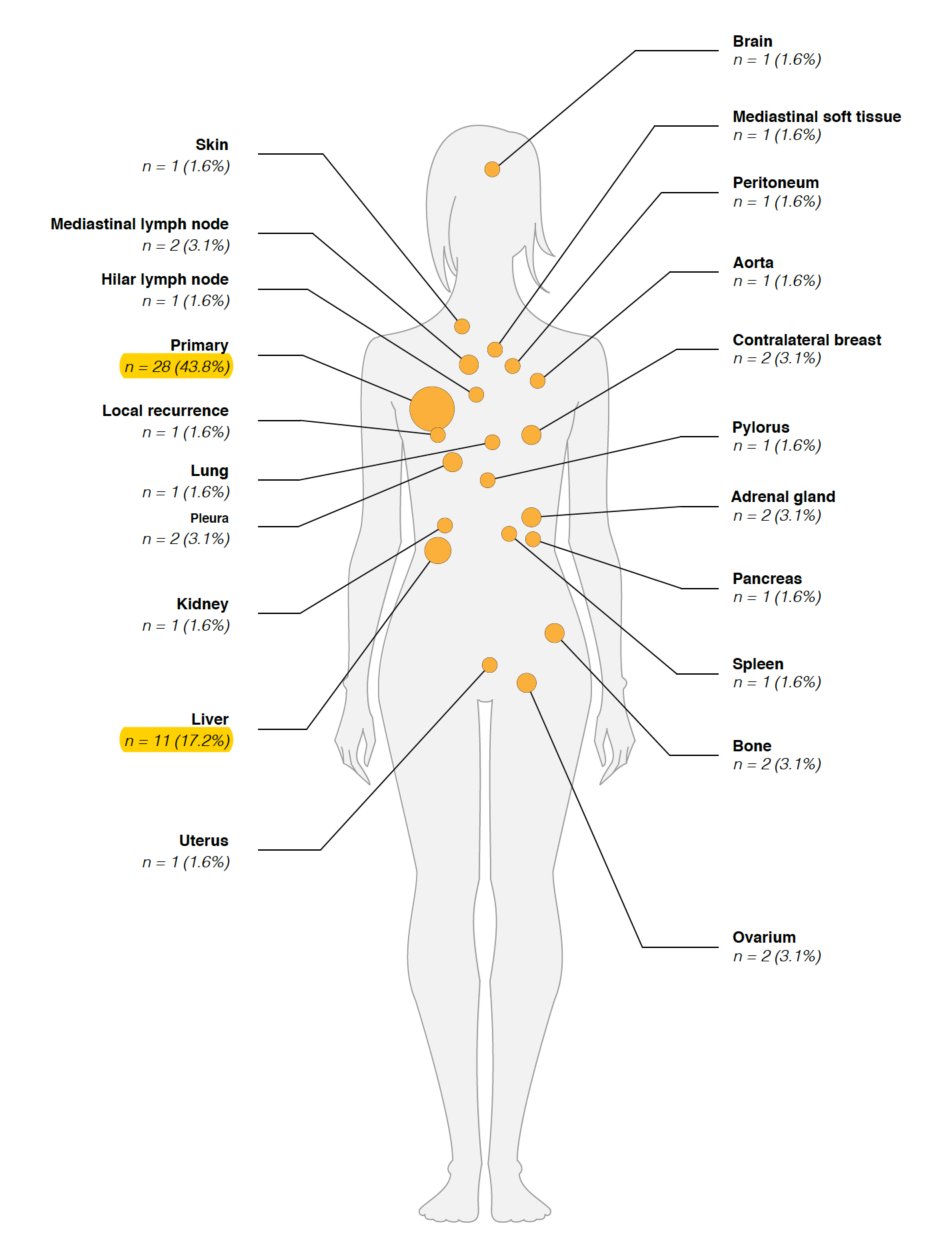
数据在:EGAS00001000760病人解剖取样
可以看到绝大部分肿瘤都会往肝脏转移
体细胞基因突变和基因拷贝数差异
两个主要结论:
- 初始诊断后不久即复发,其转移特征与原发肿瘤密切相关;
- 如果复发较晚,其转移特征与原发肿瘤的分子差异较大。
肿瘤进化
Tier-3 SNVs 构建 phylogenetic trees inferred from samples with >1500X coverage and >3% VAF are shown on the right.
Tier-4 SNVs 构建 The phylogenetic trees were inferred from samples with >30% CCF, >1500X coverage and >3% VAF.
选择不同的SNVs可以构建不同的进化树:
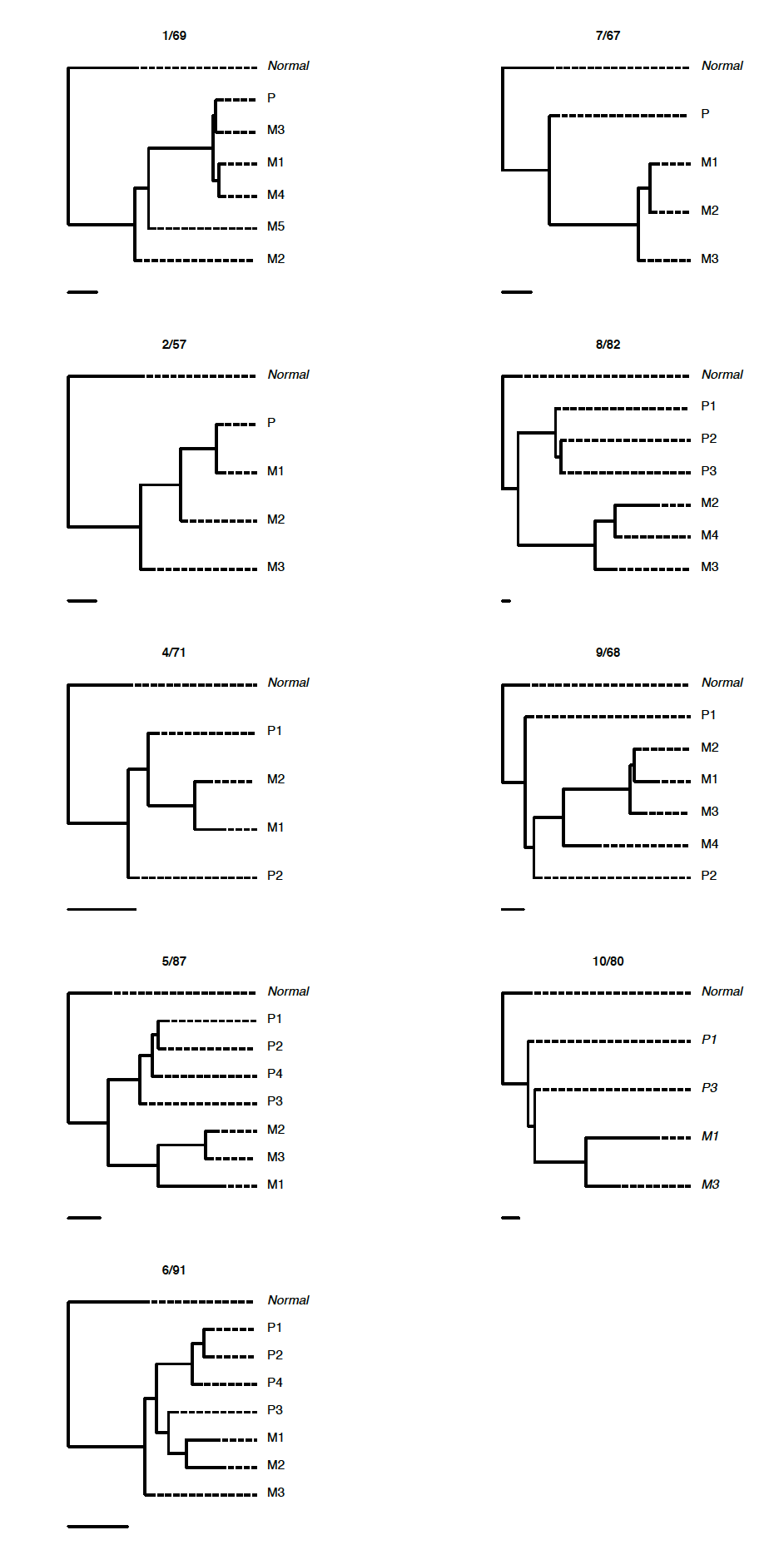
每个病人单独分析
首先是画出取样部位:
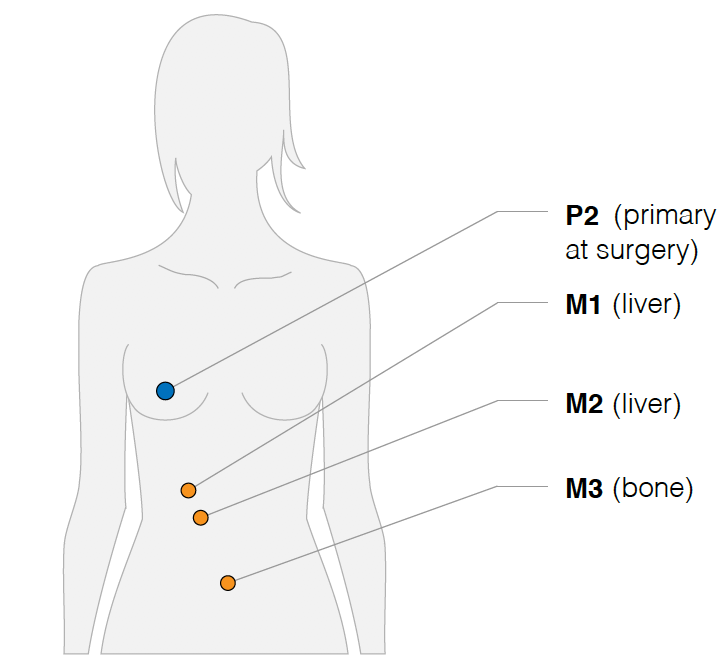
然后分析其不同部位的WES数据的somatic的SNVs共有情况,如下:
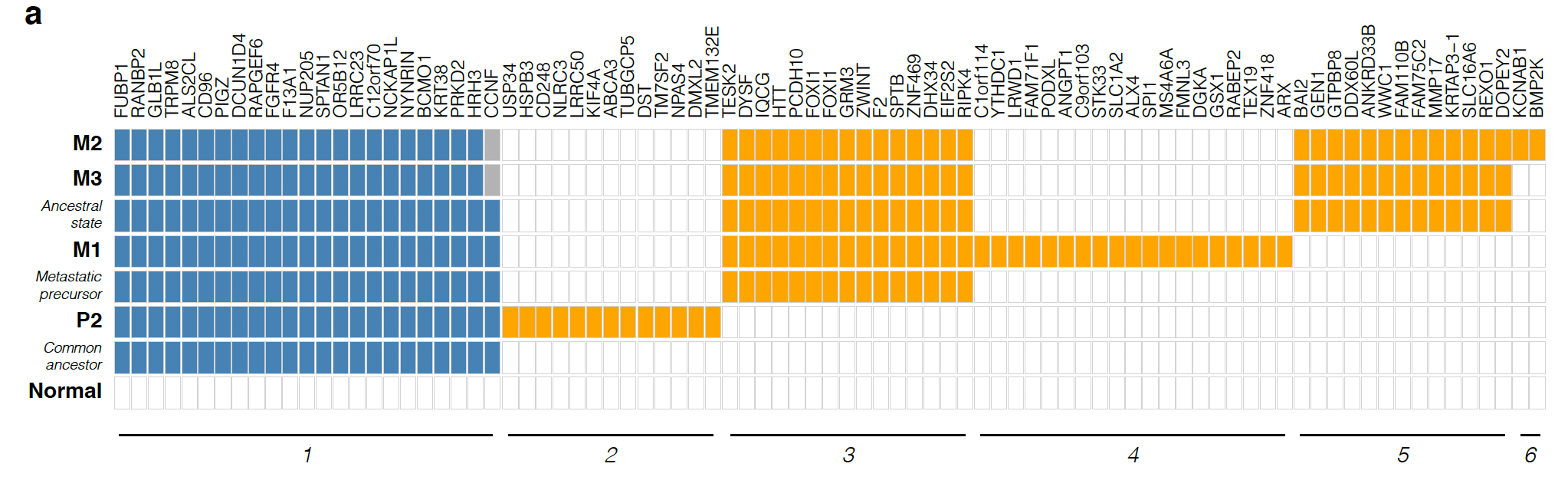
最后根据这些somatic的SNVs和CNV分别或者一起构建进化关系图:
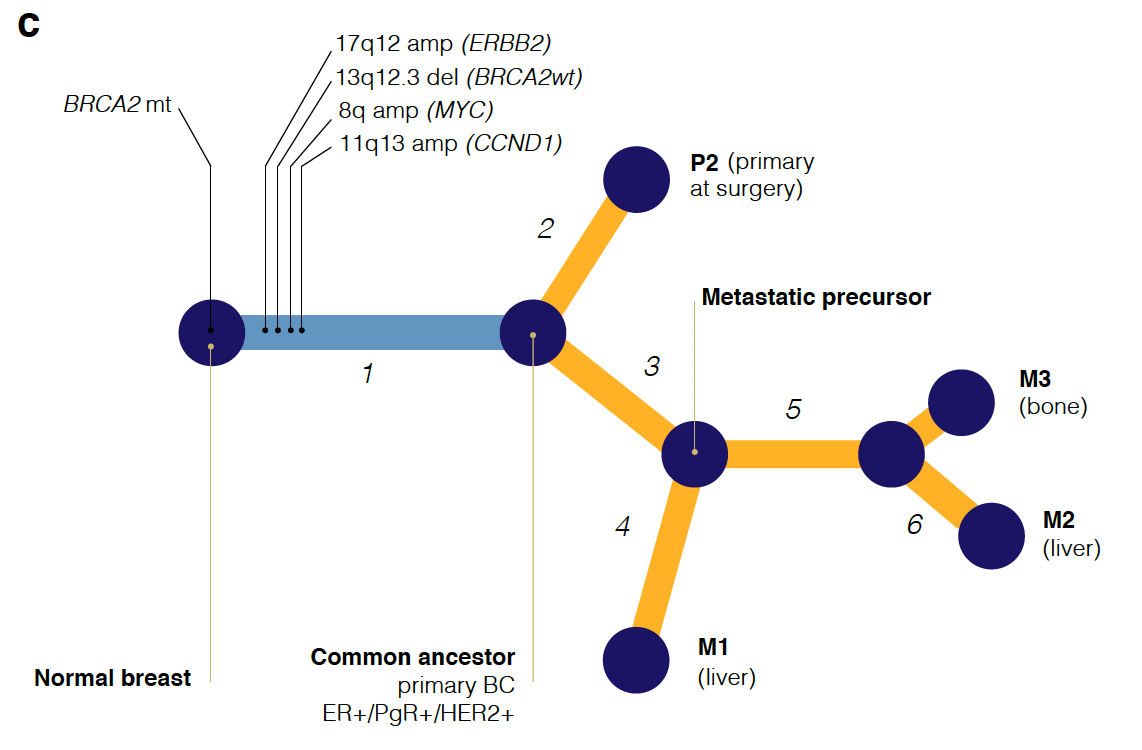
统计学知识很多
想完全看懂不容易,而且作者并没有给出原始数据,但是作者给出了自己的分析结果,就是somatic的SNVs和CNV,理论上看懂了他们的分析方法,是可以构建同样的进化关系。
- Supplementary Data 1
Patient clinico-pathological characteristics - Supplementary Data 2
Central pathological review of lesions profiled - Supplementary Data 3
Details of somatic single nucleotide variants and indels indexed by whole exome - Supplementary Data 4
Details of somatic copy number aberrations indexed by OncoScan FFPE