2433个乳腺癌患者的173个基因的突变全景图
发表于2016年的NC,The somatic mutation profiles of 2,433 breast cancers refine their genomic and transcriptomic landscapes 可以说后续做乳腺癌人群队列突变研究的都需要引用这篇文章的数据结果,里面涉及到的分析要点也比较多,都是比较容易重现的。
这2433个病人,来自于 METABRIC 计划,已经有
- copy number aberration (CNA)
- gene expression
- long-term clinical follow-up
的信息,所以这个时候再加入173个基因的捕获测序,可以更加全面的了解乳腺癌患者。
乳腺癌具有患者间与同一患者肿瘤内的基因组变异性。以患者间的异源性分类早期乳腺癌生物亚型,现在临床对乳腺癌患者通常是观察 morphological assessment (size, grade, lymph node status) ,或者检查,ER,PR,HER2 等marker,目前的亚型主要是以下: - 管腔A型(luminal A)
- 管腔B型(luminal B)
- 类正常乳腺型(normal breast-like)
- HER-2型
- 基底细胞样(basal-like)乳腺癌。
Pereiral等通过测序2433例乳腺癌样本的173个基因,发现40个肿瘤抑制基因和癌基因的驱动基因(多重驱动),这些基因参与的生物学过程包括: - AKT信号
- 细胞周期调节
- 染色质功能
- DNA损伤与凋亡
- MAPK信号
- 组织架构
- 转录调节
- 泛素化
并且发现ER+乳腺癌患者PI3K突变与不同的生存相关。实验前挑选基因
挑选的173个基因,来自于前面的TCGA计划,下面简单列出几个基因:
#Supplementary Dataset 1 - Details of genes & mutations in this study #Genes names, positions and annotation transcripts, numbers of various classs of mutations, numbers of CNAs, numbers of samples with double mutations, whether gene was included because of homozygous deletions完整表格见: Supplementary Data 1
| HGNC_symbol | Chr | Start | End | Strand | Annotation_transcript | Number_mutations | Number_synonymous | Number_missense |
| —————- | —— | ————- | ————- | ——— | ——————————- | ———————— | ————————- | ———————- |
| ACVRL1 | 12 | 52300702 | 52317645 | + | ENST00000388922 | 72 | 7 | 12 |
| AFF2 | X | 147581639 | 148082693 | + | ENST00000370460 | 296 | 28 | 40 |
| AGMO | 7 | 15239443 | 15602140 | - | ENST00000342526 | 117 | 11 | 24 |
| AGTR2 | X | 115301458 | 115306725 | + | ENST00000371906 | 40 | 0 | 14 |
| AHNAK | 11 | 62200516 | 62314832 | - | ENST00000378024 | 387 | 82 | 237 |
| AHNAK2 | 14 | 105403091 | 105445194 | - | ENST00000333244 | 878 | 322 | 524 |
| AKAP9 | 7 | 91569689 | 91740487 | + | ENST00000356239 | 265 | 30 | 137 |
| AKT1 | 14 | 105235187 | 105262580 | - | ENST00000554581 | 193 | 17 | 96 |
| AKT2 | 19 | 40735724 | 40791765 | - | ENST00000392038 | 138 | 10 | 12 |
| ALK | 2 | 29415140 | 30144932 | - | ENST00000389048 | 188 | 37 | 49 |
| APC | 5 | 112042702 | 112182436 | + | ENST00000457016 | 159 | 18 | 55 |
| ARID1A | 1 | 27022022 | 27109101 | + | ENST00000324856 | 243 | 39 | 57 |
| ARID1B | 6 | 157098564 | 157532413 | + | ENST00000346085 | 204 | 40 | 54 |
| ARID2 | 12 | 46123120 | 46302319 | + | ENST00000334344 | 159 | 29 | 36 |
| ARID5B | 10 | 63660513 | 63857207 | + | ENST00000279873 | 143 | 18 | 39 |
| ASXL1 | 20 | 30945647 | 31027622 | + | ENST00000375687 | 142 | 21 | 50 |
| ASXL2 | 2 | 25961753 | 26101812 | - | ENST00000435504 | 128 | 13 | 42 |somatic突变结果
大部分的分析资料都是在: Supplementary Information
纯粹分析结果在 : Somatic mutation calls and ASCAT segment files for 2,433 primary tumours are available at http://github.com/cclab-brca
但是原始数据是 EGAS00001001753 需要申请才能下载。
突变仍然是以 PIK3CA (coding mutations in 40.1% of the samples) and TP53 (35.4%) 为主。
其次就只有5个基因突变超过10%的样本了,分别是:MUC16 (16.8%); AHNAK2 (16.2%); SYNE1 (12.0%); KMT2C (also known as MLL3; 11.4%) and GATA3 (11.1%) ,但是MUC16 本身的背景噪音太大,不适合二代测序这个技术。病理性的germline突变情况
还是那些出名的基因作者就拿出来说了说:
- BRCA1 and BRCA2 were identified in 1.36% and 1.64% of the cohort, respectively
- 2.22% of tumours harboured pathogenic CHEK2germline mutations.
- TP53 pathogenic germline mutations were found in 0.82% of the tumours.
突变过滤策略
值得注意的是: All reads with a mapping quality < 70 were removed prior to calling.
其它策略包括: - Based on our analysis of replicates, SNVs with MuTect quality scores <6.95 were removed.
- We removed those variants that overlapped with repetitive regions
- Fisher’s exact test was used to identify variants exhibiting read direction bias
- SNVs present at VAFs smaller than 0.1 or at loci covered by fewer than 10 reads were removed, unless they were also present and confirmed somatic in the Catalogue of Somatic Mutations in Cancer (COSMIC).
- 删除那些在千人基因组计划的任意人群(AMR, ASN, AFR) 里面频率大于1%的变异位点。
- We used the normal samples in our data set (normal pool) to control for both sequencing noise and germline variants, and removed any SNV observed in the normal pool (at a VAF of at least 0.1).
这些策略理论上是需要引入到自己的研究里面的。
找driver突变
使用的是: Vogelstein et al.16 的方法 , 定位了 40个基因 , We used a ratiometric method to identify 40 Mut-driver genes
主要是区分recurrent和inactivating的突变
其中recurrent突变包括
- nonsynonymous SNVs
- in-frame indels
- oncogene score (ONC)
而inactivating突变包括: - frameshift indels
- nonsense SNVs
- splice site mutations
- tumour suppressor gene score (TSG)
The mutation patterns of some Mut-driver genes differed by ER status.
值得注意的是: - Overall, 22.6% of tumours harboured a coding mutation in one of the seven Mut-driver genes involved in chromatin function (KMT2C, ARID1A, NCOR1, CTCF, KDM6A, PRBM1 and TBL1XR1).
- Of the 40 genes, 8 were independently identified as Mut-driver tumour suppressor genes using the ratiometric method described above: FOXO3, CTNNA1, FOXP1, MEN1, CHEK2 in ER+ tumours; CDKN2A, KDM6A and MLLT4 in both ER+ and ER− tumours.
探索不同突变直接的关系,互斥或者共发生
首先是somatic的SNVs的 关系,如下图:

只要有了这些突变信息,比如maf格式的somatic mutations就可以用现成的R包,比如maftools来做上图。
然后是somatic的CNVs的关系,如下图

这个要稍微复杂一点,把拷贝数变异和点突变信息来互相联系。根据 IntClusts 分类来看突变情况
前面的分析,都是根据ER表达情况来对两千多个乳腺癌患者进行分类,现在是通过作者前面发表的 IntClusts 分类来检查突变情况,下面的这个突变全景图是整个文章的精髓:
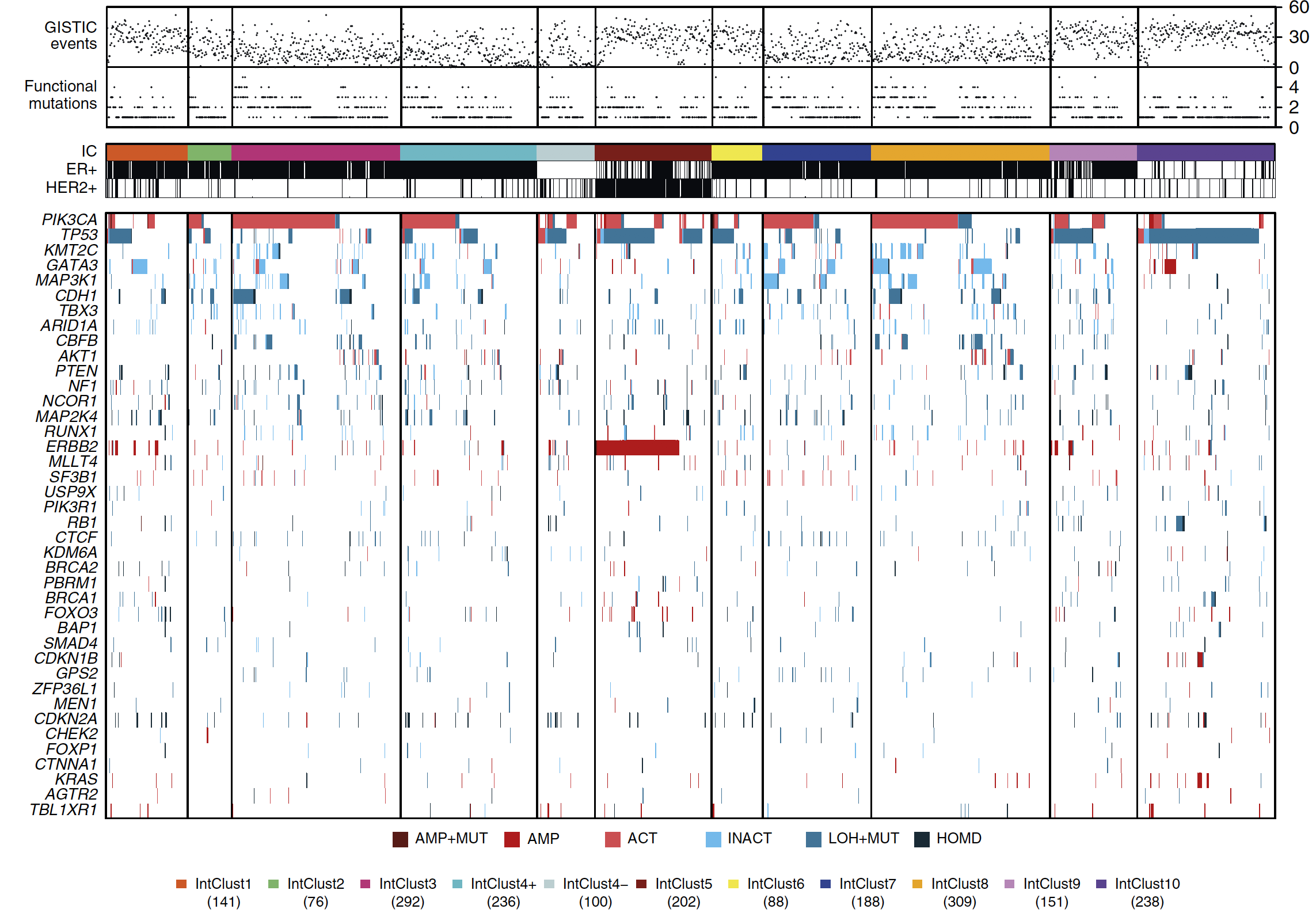
根据 mutant-allele tumour heterogeneity (MATH) 来探索肿瘤异质性
结论很清晰:
- ER+ tumours generally had lower MATH scores (median=0.29, IQR=0.18–0.44) than ER− tumours (median=0.41, IQR=0.25–0.56).
- Higher MATH scores were associated with worse outcome in ER+ cancers
这个分析也是被 maftools 包装起来了,很容易在自己的数据里面复现这个分析点。