首先声明,我不会WGCNA分析,只是大概知道它会对大量样本(>8或者15)的表达矩阵进行统计学分析,然后把表达矩阵的基因找到一下基因集合,有一些基因集合大概是非常有意义的!
因为有朋友一直好奇,我是如何学习新的知识的,所以就趁这个机会,录制了3个视频,只是我的一个学习过程而已。感兴趣可以去链接:http://pan.baidu.com/s/1jIgBTzw 密码:yh42下载,但是最后一个视频录制过程中被打断了,所以我只好重新写了个文字版的,来补充解释一下。(如果你看视频,请先看那个必看!)
学习一个新的概念,新的分析方法,我首先是谷歌了一下这个关键词,找到两个非常赞的链接!
英文的那个,让我明白了WGCNA的步骤:
就是拿到表达矩阵,根据MAD来挑选top5000个基因的表达矩阵,然后用WGCNA的包构建共表达网络,检测每一个module是什么,有什么特性。接着把这些module跟个体结合起来。
中文的那个,里面介绍了一些WGCNA的统计学原理,虽然不可能一下子看懂,但是让我大致明白它做了什么!
那么首先我视频就讲解了,如何构建表达矩阵的!
我用的是我们论坛的数据,56个breast cancer的表达矩阵: http://www.biotrainee.com/thread-603-1-1.html
然后我直接看了hope的github的代码:http://tiramisutes.github.io/2016/09/14/WGCNA.html#more
很明显,他的代码,就是总结的WGCNA 官网的tutorial而已,https://labs.genetics.ucla.edu/horvath/CoexpressionNetwork/Rpackages/WGCNA/
但是他毕竟总结了一下, 我就跟着运行一次,还不错!
- Data input and cleaning: PDF document, R script
- Network construction and module detection
- Automatic, one-step network construction and module detection: PDF document, R script
- Step-by-step network construction and module detection: PDF document, R script
- Dealing with large datasets: block-wise network construction and module detection: PDF document, R script
- Relating modules to external clinical traits and identifying important genes: PDF document, R script
- Interfacing network analysis with other data such as functional annotation and gene ontology PDF document, R script
- Network visualization using WGCNA functions: PDF document, R script
- Export of networks to external software: PDF document, R script
从代码的角度,就是上面的代码,我都在视频里面运行了,没有问题,都可以得到结果。
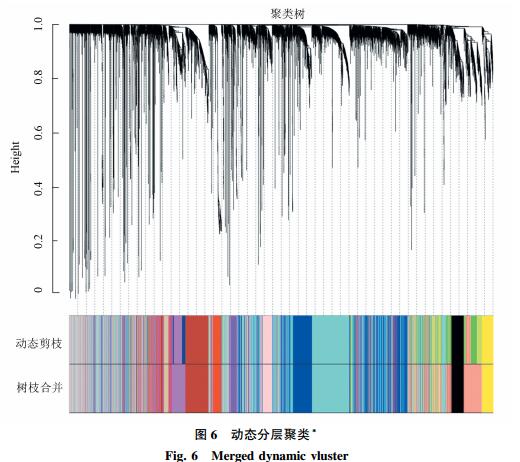
重点就是得到两个图:
#3. 一步法网络构建:One-step network construction and module detection
net = blockwiseModules(datExpr, power = 6, maxBlockSize = 6000,
TOMType = "unsigned", minModuleSize = 30,
reassignThreshold = 0, mergeCutHeight = 0.25,
numericLabels = TRUE, pamRespectsDendro = FALSE,
saveTOMs = TRUE,
saveTOMFileBase = "AS-green-FPKM-TOM",
verbose = 3)
然后是:
#1. 可视化全部基因网络 # Calculate topological overlap anew: this could be done more efficiently by saving the TOM # calculated during module detection, but let us do it again here. dissTOM = 1-TOMsimilarityFromExpr(datExpr, power = 6); # Transform dissTOM with a power to make moderately strong connections more visible in the heatmap plotTOM = dissTOM^7; # Set diagonal to NA for a nicer plot diag(plotTOM) = NA; # Call the plot function #sizeGrWindow(9,9) TOMplot(plotTOM, geneTree, moduleColors, main = "Network heatmap plot, all genes")
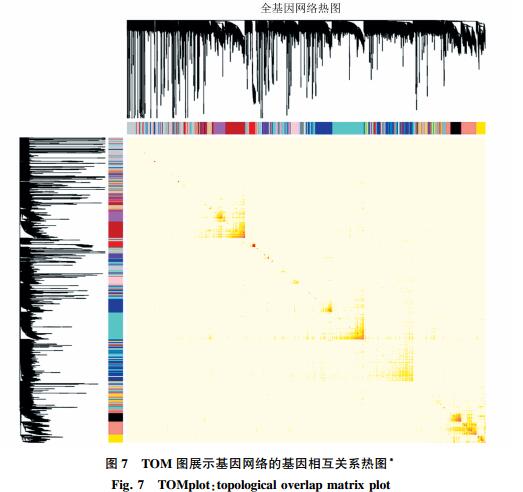
其中第二个对计算机要求比较高!
至于这些图有啥子意义,还有这些东西有多可靠,不在本次学习范围内!
其实我只是讲解了这个包如何用,能否得到那些图!下面这样的问题,我就没办法回答咯!
给群主出三个关于WGCNA的问题吧:
(1)如何确定你WGCNA得到的module所代表的共表达基因不是随机的?而确实是统计学上应该归类在那些module中的?
(2)你所用的这些样本,找到的module,鲁棒性如何?是否足够robust?
(3) 以你的breast cancer样本为例,如何证明你的modules确实可能代表乳腺癌共表达特征,如何比较他们与其他乳腺癌共表达网络的保守和差异?
(⊙o⊙)…我讲解的是如何学习WGCNA的那个包的学习方法,就是会做,会用,统计学原理我不懂啊,我也没有实战经验呀 @NJ-植物-转录组 @美国-转录组分析 如果你们是考我的话,我很抱歉了。我猜测,module的基因是否随机,看看热图,再random choose同样size的基因list看看就好了吧。 至于module是否robust,不知道WGCNA里面有没有p值的参数,没有的话,就多做几次,或者那个power换一下,比较一下。至于那些module是否代表乳腺癌共表达特征,我 更不知道了,那56个样本,是我随便找的,是就是想找一个input的表达矩阵而已,反正有了module,不都是做一些注释看看是不是合理的嘛
下面的聊天记录可能对大家的学习更有帮助!
【学神】机器猫-番茄-武汉() 12:00:19 AM
见过最多的不同rna类型用WGCNA是lnc和m miRNA和mRNA暂时还没有看过文章
【学神】中大-普外科-chaos() 12:01:40 AM
双击查看原图谢谢,我在研究研究
【学神】机器猫-番茄-武汉() 12:01:42 AM
WGCNA官网推荐 所有基因进行共表达分析 但是又有很多人说只做差异的
【学神】机器猫-番茄-武汉() 12:02:35 AM
其实我感觉 所有基因做共表达得到权重值 然后和差异的结果merge一下 貌似更好
【学神】中大-普外科-chaos() 12:02:43 AM
这个用的矩阵不是做完差异分析的normalized矩阵么?
【学神】机器猫-番茄-武汉() 12:03:10 AM
RPKM(FPKM)值 或者芯片表达量
【学神】中大-普外科-chaos() 12:03:33 AM
可能我对这个还是懵逼的吧
【学神】机器猫-番茄-武汉() 12:03:43 AM
normalized count好像不太合适吧
【学神】中大-普外科-chaos() 12:03:56 AM
counts矩阵不行么
【学神】机器猫-番茄-武汉() 12:04:10 AM
normalized count应该也可以
【学神】机器猫-番茄-武汉() 12:05:09 AM
不过不是raw count 要是normalized count
【学霸】杭州-RNA-小鸣() 12:09:05 AM
@机器猫-番茄-武汉 count数据归一化后也可以使用wgcna的
【叫兽】NJ-植物-转录组(270470585) 12:10:16 AM
应该是差异基因好做,道理上就是把变化最大的那些基因根据相关性据类,而非差异?基因变化太小,相当于引入很多噪声,直接后果是power应该会变大,因为相关性需要更高次幂才能降低噪声,出来的module不如纯用差异基因做的好
【学神】机器猫-番茄-武汉() 12:12:59 AM
说的对
【学神】机器猫-番茄-武汉() 12:13:19 AM
不过会丢掉一些相关的基因
【学神】中大-普外科-chaos() 12:14:36 AM
哎,感觉学的还是太少了,还是懂一些皮毛,光会用包跑代码不知道意义。。。
【学神】中大-普外科-chaos() 12:15:04 AM
统计学真的硬伤
【叫兽】NJ-植物-转录组(270470585) 12:15:12 AM
当然,有的表达变化1.5倍的,可能是表达调控的次级效果,但是选差异基因是就会把它排除,自然也不在共表达网络中。但是共表达网络目的就是从组学角度分清主次,抓大放小,找出?焦点
一篇中文文章也是这样做的,就是完成两个图,得到module,大多数人哪里管那么具体的统计学原理呢?

前面的4个步骤在我的学习过程中,给大家演示的清清楚楚,希望大家能get到我的思想!
后面的GO/KEGG注释我都已经讲烂了,就不赘述了!
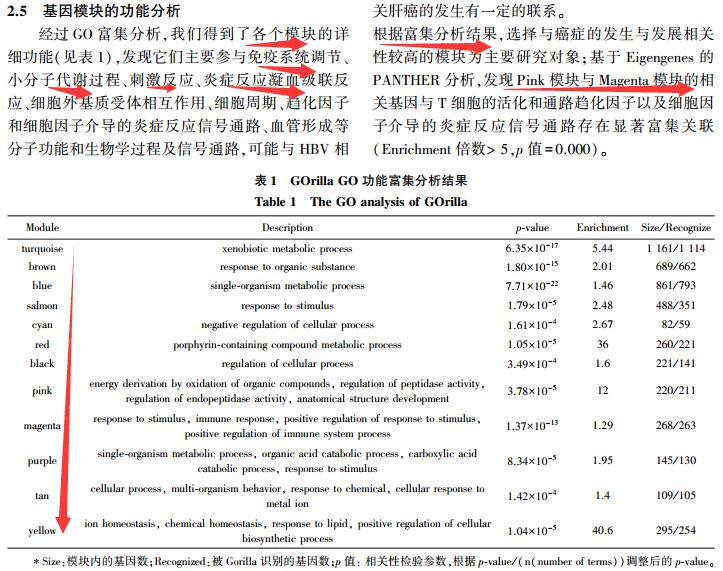
下面这个主要是网络分析的内容咯!
