一般做完一个CHIP-seq测序,如果实验设计没有问题,测序质量也OK的话,很容易了根据序列call到符合要求的peaks,或者可以去很多文章或者roadmap里面下载到非常多有意义的peaks文件, 一般是BED格式文件,这是就需要对这些peaks进行各种各样的注释以及可视化了,此时不得不强烈推荐一款网页版工具,是台湾学者开发的ChIPseek:
该工具首页就show了8张图片,就说明了该软件的功能:http://chipseek.cgu.edu.tw/index_show.py
- annotate the peaks
- link to UCSC genome browser
- provide pie charts, histograms and bar charts for peak location distribution
- apply filter criteria by peak length to get a subset of peaks
- apply filter criteria by distance to nearest TSS to get a subset of peaks
- apply filter criteria by location of the peaks
- apply filter criteria by list(s) of genes
- apply filter criteria by GO terms
- apply filter criteria by KEGG pathway annotations
- compare two datasets
- compare dataset with ENCODE transcription factor dataset
- identify enriched motif
- plot peaks on chromosome ideograms
- allow users to download figures or tables
大部分功能自己写脚本也能实现,我就不多说了。
使用方法非常简单:
然后上传自己想要分析的peaks文件
比如GSE50177里面的GSE50177_RAW.tar:http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE50177
我拿了四个peaks文件测试了一下:

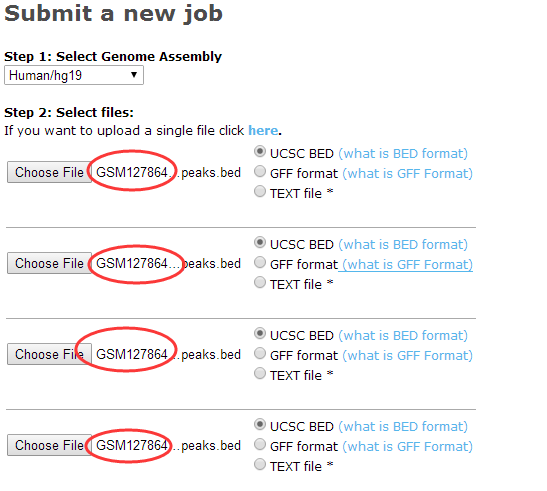
提交任务后,文件就会上传,然后网页会给一个job ID号,如果你是在一个月之内看到这篇文章,你可以直接拿我的ID号去看结果,不需要自己上传自己的文件了,当然,你肯定是需要分析自己的peaks结果的。
ChIPseek is annotating your file(s).
This page will automatically refresh every 60 seconds.
Alternatively, You may use the job ID: 1467890358.407 to visit ChIPseek latter.
一会儿就可以看到结果了,因为网页版工具的服务器容量有限,所以这个结果一个月内是有效的。
GSM1278641_Xu_MUT_rep1_BAF155_MUT (a total of 6733 peaks) (Download all annotation results)
GSM1278643_Xu_MUT_rep2_BAF155_MUT (a total of 3625 peaks) (Download all annotation results)
GSM1278645_Xu_WT_rep1_BAF155 (a total of 10987 peaks) (Download all annotation results)
GSM1278647_Xu_WT_rep2_BAF155 (a total of 5225 peaks) (Download all annotation results)
把每个文件的每个peaks都注释了,而且提供带链接的下载结果,tab分割的纯文本文件,用excel打开可能看起来舒服一点
还有4个可视化图片是我们可能会比较感兴趣的:
Peak location (pie chart)
Peak location (bar chart)
Distance to TSS
Peak length distribution
以及它可以把我们上传的bed格式peaks区域文件转为fasta序列 Peak sequences
本质是根据坐标从参考基因组里面提取序列而已,我把所有的序列都下载下来了,可以用来直接做motif查找
$ ls -lh *fasta
-rw-r–r– 1 Jimmy 197121 18M Jul 7 19:40 GSM1278641_Xu_MUT_rep1_BAF155_MUT_sequence.fasta
-rw-r–r– 1 Jimmy 197121 9.9M Jul 7 19:38 GSM1278643_Xu_MUT_rep2_BAF155_MUT_sequence.fasta
-rw-r–r– 1 Jimmy 197121 26M Jul 7 19:41 GSM1278645_Xu_WT_rep1_BAF155_sequence.fasta
-rw-r–r– 1 Jimmy 197121 14M Jul 7 19:41 GSM1278647_Xu_WT_rep2_BAF155_sequence.fasta