这个软件在TCGA计划里面被频繁使用者,用这个软件的目的很简单,就是你研究了很多癌症样本,通过芯片得到了每个样本的拷贝数变化信息,芯片结果一般是segment结果,可以解释为CNV区域,需要用GISTIC把样本综合起来分析,寻找somatic的CNV,并且注释基因信息。
有两个难点,一是在linux下面安装matlab工作环境,二是如何制作输入文件。
一、程序安装
安装指南:ftp://ftp.broadinstitute.org/pub/GISTIC2.0/INSTALL.txt
软件官网: http://www.broadinstitute.org/cgi-bin/cancer/publications/pub_paper.cgi?mode=view&paper_id=216&p=t
下载:wget ftp://ftp.broadinstitute.org/pub/GISTIC2.0/GISTIC_2_0_22.tar.gz
它的文档写的非常详细:ftp://ftp.broadinstitute.org/pub/GISTIC2.0/GISTICDocumentation_standalone.htm
解压之后,需要自己安装matlab编译环境,这个会很麻烦!
解压之后,需要自己安装matlab编译环境,这个会很麻烦!
二、输入数据准备
用picnic或者birdseed等软件处理snp6.0芯片的raw data之后得到的segment文件
多个样本的segment合并起来作为输入数据,还有样本列表,芯片的一些信息,根据示例文件,很容易做出input文件!
arraylistfile就是你本次运行GISTIC软件所涉及到的所有样本,一般一个癌种一起运行。
cnvfiles可以不用。
segmentationfile.txt 就是你snp6.0等芯片运行得到的segment信息,把所有样本的结果合并在一起,一般一个样本的segment有1000千左右
markersfile.txt主要取决于你的芯片平台,如果是affymetrix的snp6.0芯片,会有90多万行数据,每个探针的信息都有。
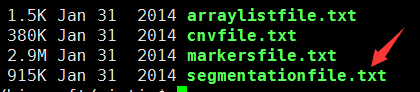
软件自带的测试数据如上,可以看到是106个样本,总共是两万多segment信息,那么也就意味着平均每个样本才200个,可能是snp6.0芯片数据的PICNIC软件的结果。但是它的
markersfile.txt 明确写着才十多万mark,也就是探针,所以应该不是
snp6.0芯片
106 arraylistfile.txt
12942 cnvfile.txt
115593 markersfile.txt
20521 segmentationfile.txt
三、程序使用
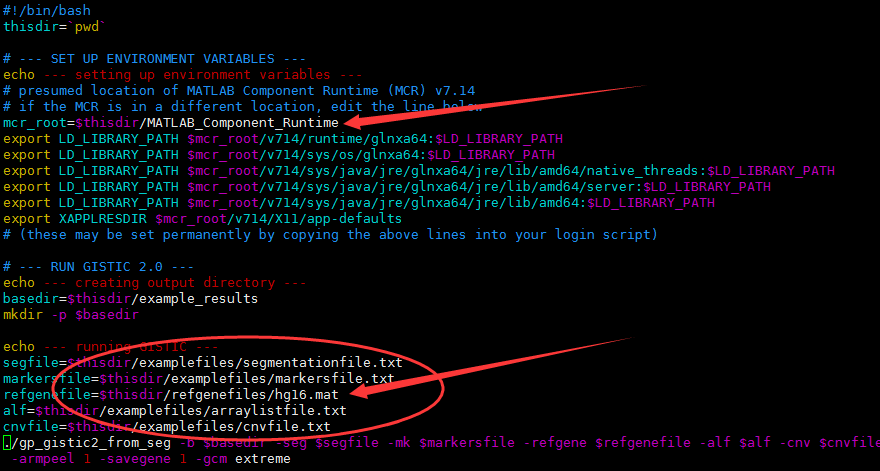
软件提供的运行脚本使用的是csh,我修改成了bash
还需要修改matlab的路径及基因组版本信息
四、输出数据解读
简单解释下输出的目录下的文件
all_data_by_genes.txt 代表了基因(包括非编码RNA如miRNA,lncRNA)在样本中具体的拷贝数值。
all_lesions.conf_90.txt 代表识别的拷贝数扩增和缺失Peak区域。
all_thresholded.by_genes.txt 代表离散化之后的数值,如-2代表丢失两个拷贝,-1代表丢失一个拷贝,0代表拷贝数正常,1代表增加一个拷贝,2代表扩增两个拷贝。
broad_significance_results.txt代表显著发生拷贝数变异的broad区域。
broad_values_by_arm.txt 代表染色体臂在样本中的拷贝数数值。
scores.gistic代表通过该方法打分之后的结果。
我写这个教程应该是2016年夏季了,现在已经是2017年秋季,这个软件又更新了,增加了对hg38版本的参考基因组数据进行处理,同时还把csh更改成了bash,真棒!
2.0.23 (2017-03-27) - The markers file input is now optional - if omitted, pseudo-markers will be
generated to satisfy GISTIC's input requirements while ensuring reasonably
uniform coverage of the genome.
- The "broad analysis" of arm-level events has been revised:
(1) arm-level events are now called from a single broad copy number profile
instead of separate amplification and deletion profiles, which had led to
arms counterintuitively called as amplified and deleted on the same sample;
(2) the frequency scores used to determine z-scores and q-values, which excludes
arms with the opposite call from the denominator, are now in a column called
"frequency score". A new column called "frequncy" gives the intuitive frequency
with the denominator inluding arms from all the samples. The analysis results
for the same data will be different from that of previous GISTIC versions.
- Error handling messages have been improved. In particular, many informative
error messages were masked by an "Index exceeds matrix dimensions" error
in the exception handler itself.
- An hg38 reference genome is included with this release.
- The gp_gistic2_from_seg binary executable is now compiled for MCR 8.3
(Matlab R2014a). The source code is compatible with versions of Matlab up to
R2016a, however, the appearance of output graphics may be altered for Matlab
versions R2015a and later.
- This release adds the convenient 'gistic2' wrapper function which sets up
the MCR and passes its command line argument to the executable. Scripts have
been converted from the C-shell to the Bourne shell.
(END)
generated to satisfy GISTIC's input requirements while ensuring reasonably
uniform coverage of the genome.
- The "broad analysis" of arm-level events has been revised:
(1) arm-level events are now called from a single broad copy number profile
instead of separate amplification and deletion profiles, which had led to
arms counterintuitively called as amplified and deleted on the same sample;
(2) the frequency scores used to determine z-scores and q-values, which excludes
arms with the opposite call from the denominator, are now in a column called
"frequency score". A new column called "frequncy" gives the intuitive frequency
with the denominator inluding arms from all the samples. The analysis results
for the same data will be different from that of previous GISTIC versions.
- Error handling messages have been improved. In particular, many informative
error messages were masked by an "Index exceeds matrix dimensions" error
in the exception handler itself.
- An hg38 reference genome is included with this release.
- The gp_gistic2_from_seg binary executable is now compiled for MCR 8.3
(Matlab R2014a). The source code is compatible with versions of Matlab up to
R2016a, however, the appearance of output graphics may be altered for Matlab
versions R2015a and later.
- This release adds the convenient 'gistic2' wrapper function which sets up
the MCR and passes its command line argument to the executable. Scripts have
been converted from the C-shell to the Bourne shell.
(END)