MAQ在2008年还是蛮火的,但是现在基本都是BWA和bowtie的天下了。
就当怀念一下它吧,给它写一个教程!
软件下载:
官网直接找到:http://maq.sourceforge.net/
我是linux系统,用wget下载:wget https://sourceforge.net/projects/maq/files/maq/0.7.1/maq-0.7.1.tar.bz2
解压,很容易观察到是C++源码,所以用源码安装三部曲来安装
tar jxvf software.tar.bz2cd software./configure --prefix=$pathmakemake test
安装之后把该软件添加到环境变量!
输入数据:
这里选择两个网络上的测试数据:
如果是真想用这个软件的话,需要参考基因组和测序数据,这个链接貌似已经年久失修啦~!
# download a test reference genome (TAIR9 Chromosome 1)
wgethttp://biocluster.ucr.edu/~tbackman/query.fastq# download some test Illumina reads from Arabidopsis
运行命令:
maq # inspect command line options
maq fasta2bfa genome.fasta genome.bfa
# create binary of reference genome
maq fastq2bfq query.fastq readBinary.bfq
# create a binary of dataset
maq match out.map genome.bfa readBinary.bfq
# align query to genome and store output
结果解读:
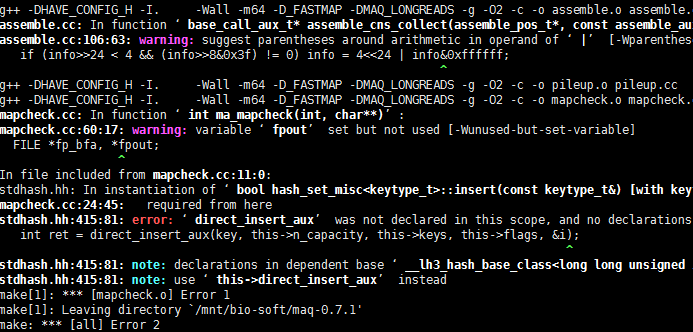
我在想,这个MAQ软件发明之前,好像还没有SAM文件格式的定义,那么它的结果
out.map肯定不是sam格式的。哈哈,这个软件我无法安装,换了好几系统也没成功,如果是太老了,很多库文件却是。我也懒得去解决了。这种报错,对我这样的非计算机专业来说,简直是天书!