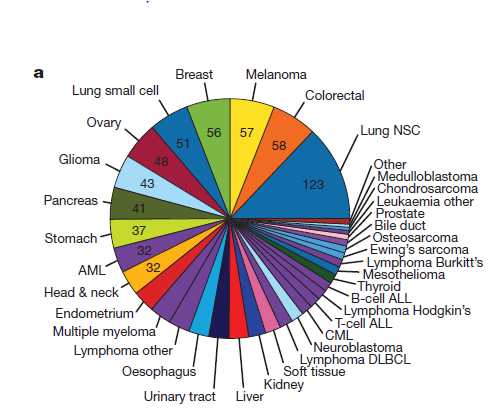
我看到这篇science的补充材料最后一个图是: Continue reading
二
14
我看到这篇science的补充材料最后一个图是: Continue reading
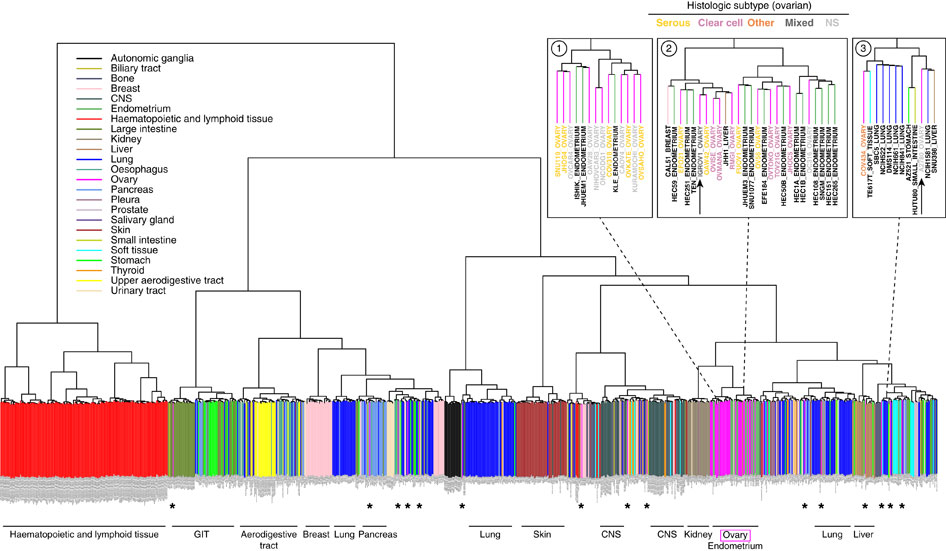
step4:Unsupervised hierarchical clustering (1-Spearman distance, average linkage) was performed on the cell lines using the aCGH data.
Putative driver genes of which copy number aberrations correlated to mRNA gene expression were identified to determine subtypes or clusters that are driven by different mechanisms. This was done using Mann Whitney U-test with p<0.05, and Spearman Correlation Coefficient test with Rho >0.6.
step5:We then performed consensus clustering[17] on the gene expression data of the 27 gastric cancer cell lines from CCLE using these putative driver genes. We selected k = 2 as it gives sufficiently stable similarity matrix.
step6: In order to assign new samples to this integrative cluster, significance analysis of microarray (SAM) [18]with threshold q<2.0 was used to generate subtype signature based on the mRNA expression data of the 1762 genes from the 27 gastric cancer cell lines in CCLE.
先用甲基化数据来聚类,得到putative driver genes,然后再用这些基因的表达数据来再次聚类,分成两类,然后对这两类进行SAM找差异基因
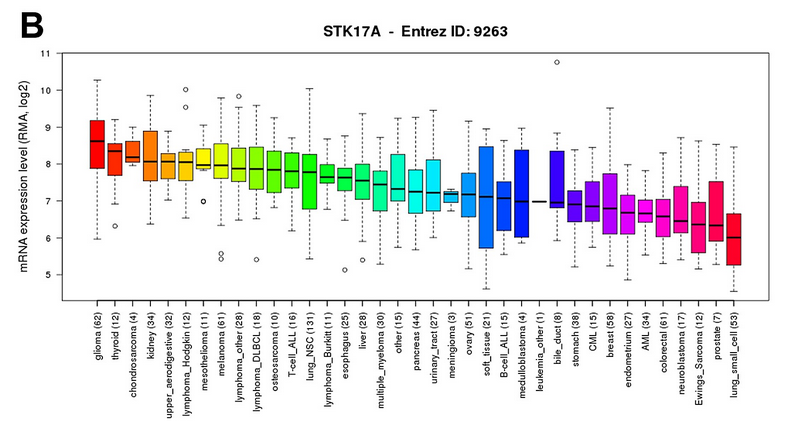
结论就是:STK17A is highly expressed in glioma cell lines compared to other cancer types. Data was obtained through the Cancer Cell Line Encyclopedia (CCLE).
第三篇文献:http://www.nature.com/ncomms/2013/130709/ncomms3126/fig_tab/ncomms3126_F4.html