我以前一直以为有了bwa跟bowtie,没什么必要用其它的alignment软件,直到我碰到了高插入删除的helicos三代测序数据,我才发现,这个古董软件genomemapper居然大有用武之地了。
一.下载并且安装该软件
这是最新版本了
| Release 0.4.4 | 2012-10-30 | source code including documentation |
Wget http://1001genomes.org/data/software/genomemapper/genomemapper_0.4.4/genomemapper-0.4.4.tar.gz
这个软件安装很简单,解压进入目录,make一下即可
看到make完了之后就会多了两个软件,其中一个是用来构建参考基因组索引,一个用来比对的!
二.准备数据
既然是比对软件,那么肯定是一个参考基因组,一个测序的fastq原始文件咯
当然这个软件比较奇葩,它还支持Multi-FASTA, FASTQ2 or SHORE flat file format,
三、比对命令
这里要分两步走,首先是构建参考基因组的索引,然后才是比对
/home/jmzeng/bio-soft/genomemapper-0.4.4/gmindex \
-i BRCA1.fa -x BRCA1.idx -t BRCA1.meta
首先构建索引,种子长度就用默认的12即可,然后构建完索引如下。
然后进行比对即可
/home/jmzeng/bio-soft/genomemapper-0.4.4/genomemapper \
-i BRCA1.fa -q SRR258835.fastq -M 4 -G 2 -E 4 -o mapped_reads.fl -u unmapped_reads.fl
成功比对的都输出到了mapped_reads.fl -这个文件,未比对上的在unmapped_reads.fl
我有12344条序列,成功比对的只有5276条,但是如果我用精确比对的算法,只有一千五百条是可以比对的,所以用这个允许4个mismatch和2个gap的比对算法,大大提高了比对率。
然后我修改了比对参数可以达到5605,5654,5696的提升。但是没有质的飞跃,估计本身我的这种helicos测序数据错误率就太可怕了。
四,输出结果解读
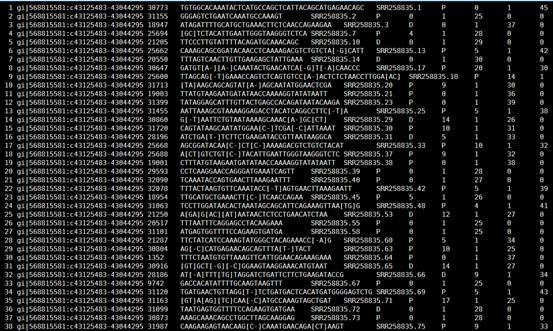
这个是很规则的tab键分割的文本字符,我就不解读了,大家看readme