太多人问到:自己想挖掘的癌症,虽然是在TCGA数据库有数据,但是normal(癌旁样品或者血液)太少了,做差异分析什么的, 会面临样本数量不平衡问题,是否可以纳入GTEx数据库的正常组织转录组测序数据。
- GTEx,The Genotype-Tissue Expression (GTEx) project,首次被提出来是2013年,上百位科学家联名在Nature Genetics杂志发表的文章首次介绍了“基因型-组织表达工程”,并成立了“基因型-组织表达研究联盟”(Genotype-Tissue Expression Consortium,GTEx)以下简称“GTEx”)。The GTEx has catalogued gene expression in >9,000 samples across 53 tissues from 544 healthy individuals.
- TCGA,The cancer genome altas,https://cancergenome.nih.gov/ ,是由National Cancer Institute ( NCI, 美国国家癌症研究所) 和 National Human Genome Research Institute (NHGRI, 国家人类基因组研究所) 合作建立的癌症研究项目,通过收集整理癌症相关的各种组学数据。The Cancer Genome Atlas (TCGA) has quantified gene expression levels in >12000 samples from >33 cancer types.
其实是没办法简单的回答是否可以整合TCGA和GTEx数据库,或者说该如何结合,这背后的统计学略微有点复杂,不仅仅是批次效应。发表在Sci Data. 2018 的文章:Unifying cancer and normal RNA sequencing data from different sources 就比较详细的说明了TCGA和GTEx数据库的转录组数据的天然差异: - sequencing platform and chemistry, personnel, details in the analysis pipeline, etc
- 基因表达量范围:4-10 (log2 of normalized_count) for TCGA, and 0-4 (log2 of RPKM) for GTEx
全部代码共享在:GitHub (https://github.com/mskcc/RNAseqDB).
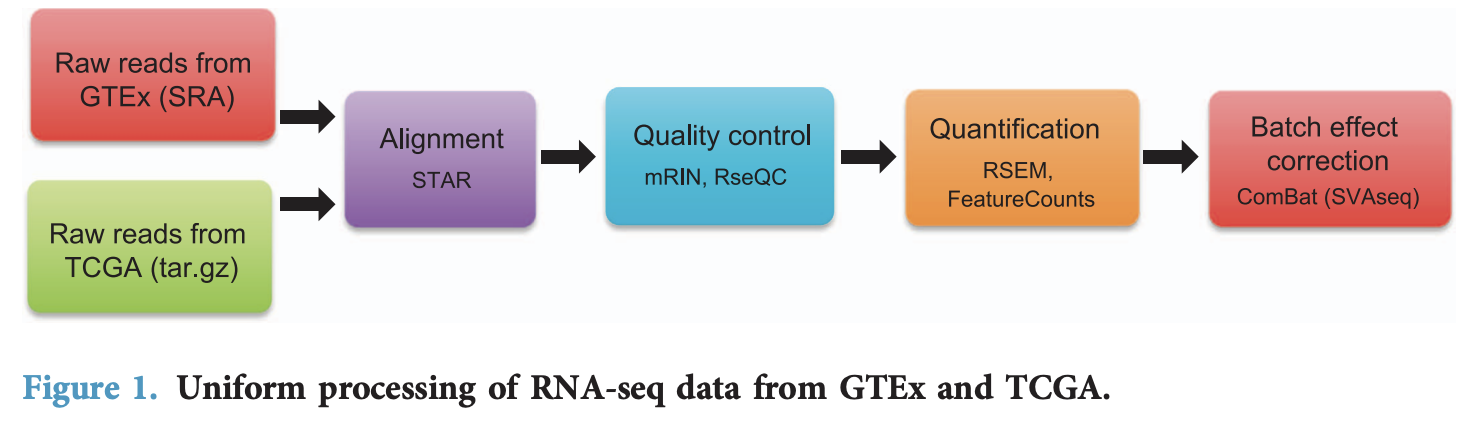
最近一篇发表在SR,17 February 2020 的文章:Variability in estimated gene expression among commonly used RNA-seq pipelines 比较了常见转录组测序数据分析流程对定量拿到的表达矩阵的影响: - We compared gene expression values from common samples (4,800 tumor samples from TCGA and 1,890 normal-tissue samples from GTEx) processed by the pipelines to understand how gene expression quantification is impacted by differences in data processing.
TCGA和GTEX是两个超级大的拥有RNA-seq数据的计划,其中TCGA涵盖33种癌症,超1万个样品,而GTEX也有500多个病人的50多种组织的近1万个样品数据。它们各自的发起单位对RNA-seq数据处理不一样,而且后续也有一些新的流程处理试图统一两个数据库的RNA-seq数据分析结果,比较出名的5个流程分别是: - TOPMed pipeline (https://github.com/broadinstitute/gtex-pipeline)
- recount2 pipeline (https://jhubiostatistics.shinyapps.io/recount/)
作者把这5个流程应用到TCGA和GTEX,得到10个不同组合的数据 - GDC (GDC-Xena/Toil, GDC-Piccolo, GDC-Recount2, GDC-MSKCC and GDC-MSKCC Batch).
- GTEx (GTEx-Xena/Toil, GTEx-Recount2, GTEx-MSKCC, GTEx-MSKCC Batch)
做了非常完善的比较,并且公布全部代码在:https://github.com/sonali-bioc/UncertaintyRNA
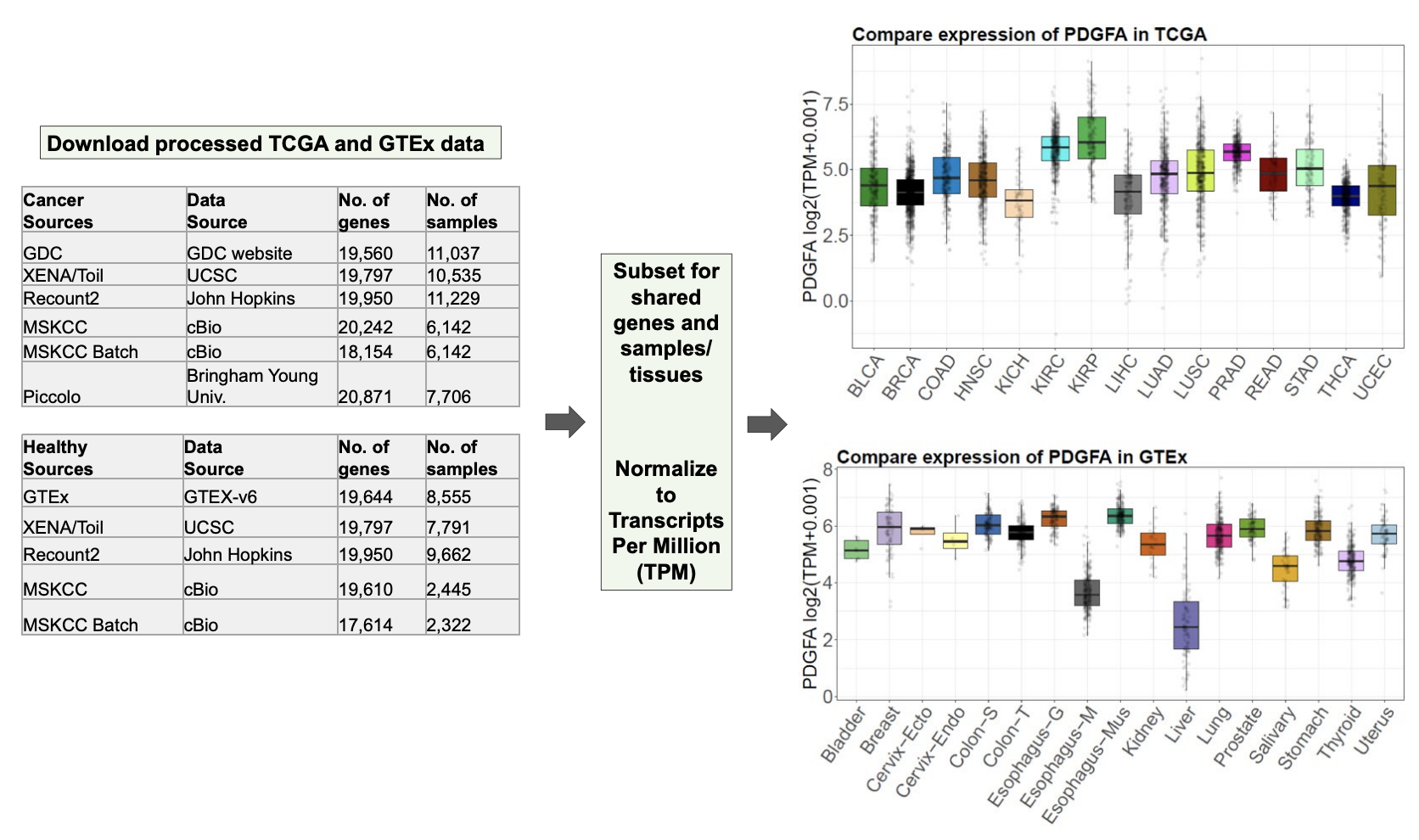
整合TCGA和GTEx数据库的文献
非常多!
很多简陋的数据挖掘,比如发表在PeerJ的 BIOINFORMATICS AND GENOMICS杂志的文章:Identification of four hub genes associated with adrenocortical carcinoma progression by WGCNA 也会涉及到TCGA数据库和GTEx的整合。
首先下载TCGA和GTEx数据库的TPM表达矩阵:
Gene transcripts per million (TPM) data were downloaded from the UCSC Xena database, which included ACC (The Cancer Genome Atlas, n = 77) and normal samples (Genotype Tissue Expression, n = 128).
然后差异分析流程是: - Of the 60,498 genes in each sample, we removed genes with a mean TPM ≤ 2.5 (>1 is a common cutoff for determining if an isoform is expressed or not in the cancer and normal samples and thus retained 13,987 genes.
- For those genes in the samples that showed significant changes, we used analysis of variance (ANOVA) in R to determine the variance in genes between the two groups. ANOVA is a collection of statistical models useful for DEG analysis.
- We obtained 2,953 significant DEGs (Table S2) in ACC with a p < 0.001 and |log2 (fold-change)| > 1 cutoff.
差异分析结果是:1,181 up-regulated and 1,772 down-regulated genes.
可以看到,作者默认TPM这个转录组测序表达数据归一化形式本身是具有跨平台跨数据库的特性,所以无需考虑批次效应,直接使用最简单粗暴的ANOVA检验即可!如果是甲基化数据
我们都知道,TCGA数据库是目前最综合最全面的癌症病人相关组学数据库,包括:
- DNA Sequencing
- miRNA Sequencing
- Protein Expression array
- mRNA Sequencing
- Total RNA Sequencing
- Array-based Expression
- DNA Methylation
- Copy Number array
知名的肿瘤研究机构都有着自己的TCGA数据库探索工具,比如: - Broad Institute FireBrowse portal, The Broad Institute
- cBioPortal for Cancer Genomics, Memorial Sloan-Kettering Cancer Center
对转录表达这个层面的信息来说,最优选择当然是整合TCGA和GTEx数据库,但是对于甲基化数据,我们有没有类似于GTEx数据库的超级大队列呢?
目前我还没有接触到,我前面分享过:这样的诊断模型才优秀,作者就是下载TCGA的结直肠癌甲基化位点信号矩阵文件: - Tissue DNA methylation data were obtained from the TCGA (TCGA, TCGA-COAD, and TCGA-READ).
以及正常人的血液的甲基化信号值作为对照: - Whole-blood DNA methylation profiles from healthy donors were generated in an aging study (GSE40279)
上面的两个队列是为了确定直肠癌特异性甲基化位点,做的是差异分析,确定了 top 1000 methylation markers
可以合理的推测应该是没有人类各个正常组织的甲基化数据供使用,所以他们才会退而求其次使用正常人的血液的甲基化信号值作为对照吧!一个开放性问题
多种分子(mRNA,lncRNA,miRNA,甲基化,蛋白)水平,它们各自的组织特异性如何?
换个方式提问就是:如果做单细胞亚群,哪个层面的信息更容易区分不同组织来源的细胞?是mRNA的表达量吗,单细胞转录组足够吗?