学员群里有小伙伴提出来了一个非常“个性化”的单细胞转录组数据分析需求:

其实几年前我也写过这样的教程,就是哪怕是同一个单细胞数据里面的不同细胞亚群质量控制参数也可以不一样,当时解读的单细胞文章是标题是:《Single-cell transcriptomics reveal the heterogeneity and dynamic of cancer stem-like cells during breast tumor progression》,因为作者发现上皮细胞比免疫细胞有更多的检测到的基因,细胞文库大小 :所以作者给出来的过滤参数不一样:
- 上皮细胞是:“total counts: > 5,000; number of genes: >2,000; the proportion of mitochondrial gene counts: <8%”
- 免疫细胞是:“total counts: >1000; number of genes: >500; the proportion of mitochondrial gene counts: <8%”
不确定是不是这样教程让大家误解了,以至于后面的数据分析大家都想模仿一下,必须给不同单细胞亚群不同的质量控制和过滤参数。
当然了,我并不是上面的学员提出来的生信分析里面的思路是完全错误,我仅仅是表面这样的需求其实是有问题的。不过不影响我们代码层面实现它,首先使用如下所示的代码载入测试数据:
# install.packages('devtools')
# devtools::install_github('satijalab/seurat-data')
library(SeuratData) #加载seurat数据集
library(Seurat)
getOption('timeout')
options(timeout=10000)
# InstallData("pbmc3k")
data("pbmc3k")
sce <- pbmc3k.final
colnames(sce@meta.data)
table(sce$seurat_annotations)
然后,针对前面的测序数据,看看不同单细胞亚群的线粒体含量,文库大小的分布情况:
## Identification of mithocondrial genes
sce[["percent.mt"]] <- PercentageFeatureSet(sce, pattern = "^MT-")
table(sce$seurat_annotations,sce$percent.mt > 0.01)
table(sce$seurat_annotations,sce$nFeature_RNA < 500 )
# ## Filtering cells following standard QC criteria.
# pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 &
# percent.mt < 5)
如下所示,很明显是可以看得到 Naive CD4 T 和 CD14+ Mono 以及B细胞是有很多细胞的数量的文库是偏小的(参数:nFeature_RNA < 500 ):
FALSE TRUE
Naive CD4 T 626 71
Memory CD4 T 482 1
CD14+ Mono 421 59
B 310 34
CD8 T 262 9
FCGR3A+ Mono 160 2
NK 154 1
DC 32 0
Platelet 4 10
这个时候学员其实是想针对性的过滤,比如仅仅是去除Naive CD4 T 里面的文库是偏小的(参数:nFeature_RNA < 500 )细胞,但是需要放过 CD14+ Mono 以及B细胞里面的。
其实过滤细胞并不需要使用 subset 函数,因为对象本质上是可以通过取子集的方式,就是按照行或者说按照列过滤即可。
kp1=sce$nFeature_RNA < 500
kp2=sce$seurat_annotations == 'Naive CD4 T'
table(kp1 & kp2)
sce_f=sce[,kp1 & kp2]
DimPlot(sce)+DimPlot(sce_f)
可能是 我的可视化技巧不够,或者说是举例的这个数据集的特性,没怎么看到过滤前后的效果对比:
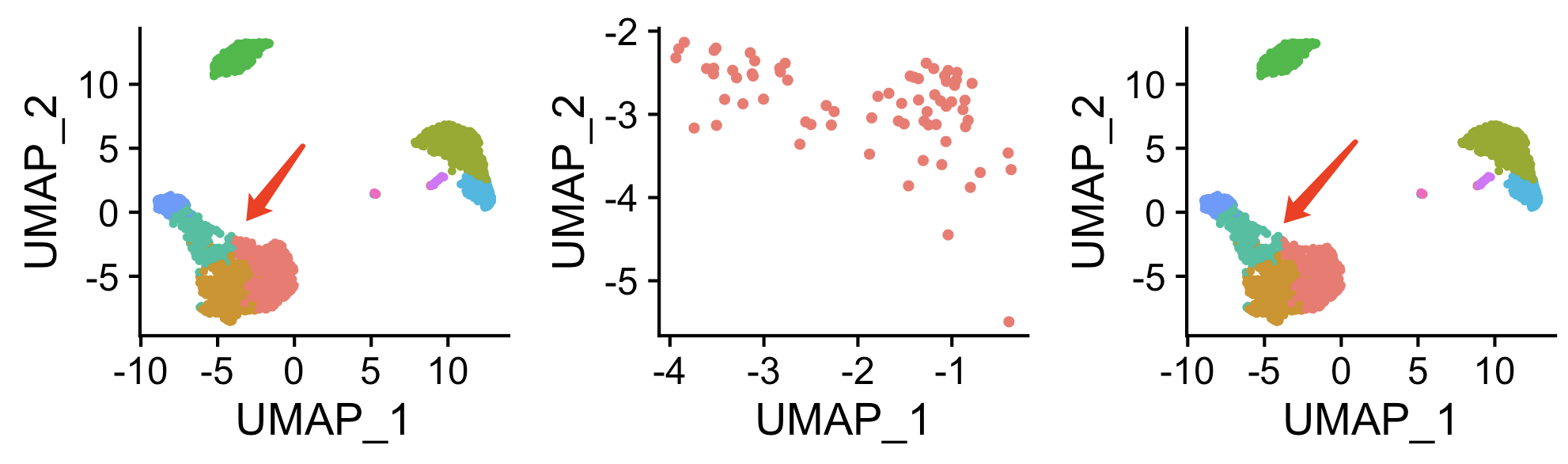
也就是说,上面的代码就实现了仅仅是针对去除Naive CD4 T 里面的文库是偏小的(参数:nFeature_RNA < 500 )细胞,去除了71个细胞,而不会影响其它单细胞亚群的文库偏小的细胞,也没有影响前面的降维聚类分群后的全部的PCA等信息。
但是,这个思路是否值得采取,是有待商榷的哦。另外,号召大家参与讨论一个问题,生信分析里面的思路错误和代码错误哪个更可怕?
比如10x技术的单细胞转录组去除双细胞问题
在单细胞转录组分析中,双细胞(Doublets)是指在同一个细胞单元中同时捕获到两个不同的细胞的情况。双细胞可能会对单细胞转录组数据的解释和分析造成困扰,因此在分析过程中通常大家看到的教程都会高速大家去除它们 :
- 技术偏差: 单细胞转录组技术在分析过程中可能引入一些技术偏差,例如RNA测序深度不均一、细胞溶解程度不同等。当双细胞形成时,两个细胞的RNA会混合在一起,可能会导致更大的技术偏差,影响后续的数据分析。
- 噪音增加: 双细胞中的RNA来自两个不同的细胞,其基因表达情况可能会相互干扰,增加数据中的噪音。这会对单细胞转录组数据的解释和生物学结论造成不确定性。
- 数据解释困难: 在双细胞中,两个不同细胞的基因表达可能会混淆,使得数据的解释变得复杂。双细胞存在的情况下,很难准确地分析每个细胞的特征和功能。
- 分析结果失真: 如果不去除双细胞,分析结果可能会受到影响,例如聚类、差异基因分析等。双细胞可能会在聚类中形成混杂的簇,使得单细胞群体结构的解释变得困难。
- 数据干净性: 去除双细胞可以提高数据的干净性和准确性,有助于后续的生物学解释和结论。
为了去除双细胞,研究人员可以使用一些专门的算法和工具,这些算法通常基于基因表达的特征,通过计算细胞之间的相似性来识别双细胞。一些方法还可以通过模拟或模型来预测双细胞的形成概率。去除双细胞的过程可以在分析流程的早期进行,以确保后续的分析结果更准确可靠。
目前主流的两个工具是:
1. DoubletFinder:
- 优点:DoubletFinder是一种基于PCA(主成分分析)的方法,通过检测细胞在PCA空间中的位置来鉴定双细胞。它能够识别出混合细胞,对于大规模数据适用,支持不同分析平台的数据。
- 缺点:在某些情况下,可能会将真实双细胞误分类为非双细胞。
2. Scrublet:
- 优点:Scrublet是一种使用模拟方法的工具,它通过比较每个细胞的原始数据和模拟数据来鉴定双细胞。可以检测出低频率的双细胞,且对分析平台不敏感。
- 缺点:可能对数据的噪声敏感,对于少数双细胞可能会有误报。
很多人会首先把多个样品合并后,作为一个整体去走上面的两个软件,其实思路就是错误的。另外,很多人的单细胞转录组数据其实没有必要去除双细胞,摸索这个流程徒增烦劳。