生物信息学领域涉及到大量的不同种类的数据的分析和处理工作,因此这个领域就必然产生许多不同类型的软件工具,比如处理DNA、RNA、蛋白质序列等不同层面的数据。但是我们这里并不想按照组学种类来对生物信息学软件工具进行分类,因为不同组学经常是有软件是交叉的,比如fastqc软件就可以针对不同ngs组学数据进行质量控制。我这里把生物信息学软件工具按照使用难易程度的大致分成3类:
- 网页工具(最易上手)
- 云平台(有门槛,比如需要看视频教程)
- 编程语言(起码三五个月的学习)
其中网页工具和云平台都不是针对专门的生物信息学工程师设计的,因为并不需要使用者会编程语言,所以使用起来非常简单。下面来一一介绍一下它们:
网页工具(最易上手)
生物信息学领域有许多在线工具和资源,这些工具提供了各种分析和可视化功能,无需用户进行大量的本地安装和配置。而且绝大部分都是大机构开发和维护,知名度比较好的大机构包括:
- NCBI(National Center for Biotechnology Information)
- UCSC( University of California, Santa Cruz )
- EMBL-EBI(European Bioinformatics Institute)
它们各自提供了海量的工具,比如NCBI的在线blast,UCSC 的 Genome Browser,以及EMBL-EBI的EMBOSS。
当然了,网页工具其实也可以按照功能来进行分类,或者ngs组学数据种类分类就更复杂了。比如实现生物学功能数据库(go以及kegg)注释(富集分析)的就有 3大在线分析工具:Enrichr、WebGestalt、gprofiler ,还有大名鼎鼎的DAVID(Database for Annotation, Visualization, and Integrated Discovery),以及后起之秀 Metascape,它们都可以用于功能富集分析的工具,帮助理解一组基因的生物学含义。
云平台(有门槛,比如需要看视频教程)
生物信息学云平台提供了在线的计算和数据存储服务,使研究人员能够在云环境中进行生物信息学数据分析和处理,而无需购买和维护本地服务器。云平台其实很难跟上面的网页工具完全区分开,它们在使用难易程度,能解决的问题复杂程度是各有侧重点的。其中网页工具很少做存储和计算,主打的是查询,而且偏重于实现某个功能需求即可。而云平台呢,是可以系统性完成多个任务需求,比如主打单细胞全流程数据分析,或者集成了几十个甚至上百个统计可视化工具。相比较而言,云平台成本会比较高,所以很难是完全免费,一般来说可以做到对指定客户免费。
海外知名云平台
- Galaxy生信云平台 : https://usegalaxy.org/
- Seven Bridges Genomics :https://www.sevenbridges.com/
国内商业公司云平台
排名不分先后,我是在搜索引擎随意检索的,而且这些云平台因为是网页工具提供,所以很容易变化域名甚至直接倒闭:
- http://sangerbox.com/home.html
- https://cloud.oebiotech.cn/task/
- https://www.xiantaozi.com/
- https://www.biocloud.net/
- http://www.cloudtutu.com/#/login
- https://www.omicshare.com/
- http://www.bioinformatics.com.cn/
- http://cloud.vazyme.com:83/
- http://www.biocloudservice.com/home.html
编程语言(需要系统性学习计算机基础知识)
正经的生物信息学数据处理工程师通常都有跟成百上千个不同编程语言开发的生物信息学软件工具做斗争的噩梦般的经历,虽然说现在有了conda这样的软件工具管理方案,而且不同操作系统本身也有自己的软件管理方案有时候也不错,所以现在大家其实很少接触需要从零开始(从源代码)安装的软件啦。但是还是有必要了解一下不同编程语言的源代码安装软件方式,因为绝大部分操作系统默认自带了C语言编辑器,java运行环境,perl和Python默认环境,所以我们就不讲解它们本身的安装啦。
首先需要在什么是基于编程语言的生物信息学软件这个概念达成共识!
我这里给出来一点自己的看法,所谓基于编程语言的生物信息学软件,就是可以在Linux等操作系统的终端,通过命令行方式并且设置合理的参数(比如输入输出文件路径或者文件名)的命令。比如:
- Bedtools 用于处理基因组坐标(BED、GFF、VCF 等)文件,进行集合运算、区域检索和统计等操作。
- Vcftools 用于处理和分析 VCF(Variant Call Format)文件,进行变异分析和统计。
- Samtools 主要用于处理 BAM/SAM 格式的测序数据,进行排序、索引、统计和格式转换。
- DeepTools 提供了用于分析测序数据的一套工具,包括绘图、标准化、差异分析等。
它们都是在通过命令行方式并且设置合理的参数(比如输入输出文件路径或者文件名)的命令,如下所示:
bedtools intersect -a file1.bed -b file2.bed > output.bed
vcftools --vcf input.vcf --freq --out output
samtools sort -o output.bam input.bam
plotHeatmap -m matrix.gz -out output.pdf
而这个命令就有可能是不同编程语言开发的,有不同的安装方式啦,这个也是难道大部分生物信息学初学者的知识点。
C语言体系源代码
因为现在conda用得多,所以我已经很少接触C语言体系源代码软件安装啦,首先需要配置一下环境变量,另外就是configure, make, 和 make install 是在源代码中常见的一套构建和安装过程。这一流程通常被用于开源软件,使得软件在多种环境(不同的操作系统)中能够更容易地进行构建和安装。
mkdir -p $HOME/biosoft/myBin
echo 'export PATH=$HOME/biosoft/myBin/bin:$PATH' >>~/.bashrc
source ~/.bashrc
mkdir -p $HOME/biosoft/cmake
cd $HOME/biosoft/cmake
wget http://cmake.org/files/v3.3/cmake-3.3.2.tar.gz
tar xvfz cmake-3.3.2.tar.gz
cd cmake-3.3.2
./configure --prefix=$HOME/biosoft/myBin
make
make install
主要是靠make和cmake工具,接下来可以安装大名鼎鼎的samtools,代码如下所示:
## Download and install samtools
## http://samtools.sourceforge.net/
## http://www.htslib.org/doc/samtools.html
mkdir -p $HOME/biosoft/samtools
cd $HOME/biosoft/samtools
wget https://github.com/samtools/samtools/releases/download/1.3.1/samtools-1.3.1.tar.bz2
tar xvfj samtools-1.3.1.tar.bz2
# 这个压缩包下面有两个软件其实,都需要各自独立的安装
cd samtools-1.3.1
./configure --prefix=$HOME/biosoft/myBin
make
make install
$HOME/biosoft/myBin/bin/samtools --help
$HOME/biosoft/myBin/bin/plot-bamstats --help
cd htslib-1.3.1
./configure --prefix= $HOME/biosoft/myBin
make
make install
~/biosoft/myBin/bin/tabix
因为配置好了环境变量,所以理论上上面的这些软件安装后也是可以直接运行即可。但是上面的软件源代码托管的网页一般来说都是海外的,所以其实下载速度对中国大陆地区的小伙伴来说其实非常的不友好。
不过,因为有了conda这样的软件工具管理方案,所以其实C语言体系源代码一个软件的出场率就很低了,大家了解一下即可。
类似的C语言体系源代码软件安装策略都是下载解压然后configure+make+make install即可,比如:
## Download and install samstat
## http://samstat.sourceforge.net/
## http://www.htslib.org/doc/samtools.html
mkdir -p $HOME/biosoft/samstat
cd $HOME/biosoft/samstat
wget http://liquidtelecom.dl.sourceforge.net/project/samstat/samstat-1.5.tar.gz
tar zxvf samstat-1.5.tar.gz
cd samstat-1.5
./configure --prefix= $HOME/biosoft/myBin
make
make install
~/biosoft/myBin/bin/samstat --help
- configure:
configure是一个脚本,用于检查系统环境和配置选项。它会检查编译器、库、头文件等,以确保软件能够在当前环境中正确编译。configure脚本会生成一个Makefile,该文件包含了编译和链接的规则。
- make:
make是一个构建工具,用于根据Makefile文件中的规则编译源代码。make会根据依赖关系和规则,只重新编译需要更新的文件,从而提高构建效率。通常,make后面会跟着一个目标,比如make all或者只是make,用于构建软件的可执行文件。
- make install:
make install用于将已编译的二进制文件、库、头文件等安装到系统的指定位置。这个步骤需要足够的权限,因为它通常会将文件复制到系统目录(如/usr/bin、/usr/lib等)中。
这种构建和安装的流程的好处有:
- 平台独立性:通过
configure阶段,软件可以检查和适应不同的平台和环境。这使得用户能够在各种操作系统和硬件平台上使用相同的源代码进行构建。 - 定制化:通过配置选项,用户可以根据需要定制软件的构建和安装行为。例如,可以选择安装目录、启用或禁用某些功能等。
- 自动化:
make工具允许自动化构建过程,只构建修改过的文件,提高了构建效率。
请注意,对于某些软件,也可能会有其他构建工具和流程,不一定都使用 configure、make 和 make install。这一套流程的广泛使用主要是因为它简单、通用,并且在许多情况下都能够满足构建和安装的需求。
更多软件这里就不一一举例啦,每个软件都有自己的官方文档,其实看看官方文档就明白了它的最佳安装方式啦。
Java编程语言的软件
一般来说,我们的操作系统里面肯定是有Java编程语言的运行环境,所以它相关的软件其实下载解压(.jar后缀的文件)即可使用啦。
mkdir -p $HOME/biosoft/snpeff
cd $HOME/biosoft/snpeff
## http://snpeff.sourceforge.net/
## http://snpeff.sourceforge.net/SnpSift.html
## http://snpeff.sourceforge.net/SnpEff_manual.html
wget http://sourceforge.net/projects/snpeff/files/snpEff_latest_core.zip
unzip snpEff_latest_core.zip
## java -jar snpEff.jar download GRCh37.75
## java -Xmx4G -jar snpEff.jar -i vcf -o vcf GRCh37.75 example.vcf > example_snpeff.vcf
使用的时候需要在软件前面加上 java -jar 这样的方式调取不同的软件哦(.jar后缀的文件)
mkdir -p $HOME/biosoft/trimmomatic
cd $HOME/biosoft/trimmomatic
## http://www.usadellab.org/cms/?page=trimmomatic
## http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic/TrimmomaticManual_V0.32.pdf
wget http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic/Trimmomatic-0.36.zip
unzip Trimmomatic-0.36.zip
java -jar ~/biosoft/Trimmomatic/Trimmomatic-0.36/trimmomatic-0.36.jar -h
大家很难在Java编程语言的软件安装上面遇到困难。可能是比较麻烦的是jre的版本问题,Java Runtime Environment(JRE)是 Java 程序的运行环境,它包含了 Java 虚拟机(JVM)和一些运行时库。Java 应用程序在用户计算机上运行时需要安装相应版本的 JRE。比如fastqc软件就有要求的最低jre版本,FastQC 是一个用于质量控制检查的工具,它是使用 Java 编写的。要运行 FastQC,必须在计算机上安装 Java 运行环境(Java Runtime Environment,JRE)。
broad研究所也是喜欢使用Java语言开发生物信息学软件,比如大名鼎鼎的GATK系列软件套件。
Perl编程语言的软件
早期的生物信息学工具和脚本通常是用 Perl 编写的。这一传统使得很多后续的工具和脚本仍然选择 Perl,所以大家也很难完全避开perl相关软件。而且绝大部分Perl编程语言开发的软件本质上其实就一个脚本,只不过是里面的依赖于大量的Perl早期模块,所以安装Perl编程语言的软件的难点其实是在Perl模块的安装:
## Download and install homer (Hypergeometric Optimization of Motif EnRichment)
## // http://homer.salk.edu/homer/
## The commands gs, seqlogo, blat, and samtools should now work from the command line
mkdir -p $HOME/biosoft/homer
cd $HOME/biosoft/homer
wget http://homer.salk.edu/homer/configureHomer.pl
perl configureHomer.pl -install
perl configureHomer.pl -install hg19
更麻烦的perl模块的安装,装Perl模块有两种方法
- 自动安装 (使用CPAN模块命令来自动完成下载、编译、安装的全过程)
- 手工安装 (去CPAN网站下载所需要的模块,手工编译、安装)
这个是另外的话题了,感兴趣的可以阅读:http://www.bio-info-trainee.com/2451.html
基于Python编程语言的软件
Python是生物信息学领域的后起之秀,基于Python开发的软件数量早就远超于传统的Perl啦,目前多组学的ngs流程里面也有很多步骤的软件是Python开发的。
单个模块就是软件
比如MultiQC,它 可以接受来自各种生物信息学工具的输出文件,包括但不限于质控工具(例如 FastQC)、比对工具(例如 BWA、Bowtie)、RNA-seq 工具(例如 STAR、HISAT2)、ChIP-seq 工具等。通过整合这些工具的输出,MultiQC 可以提供全面的数据质量和分析结果。MultiQC 能够自动检测当前目录下的分析结果文件,并生成一个 HTML 报告。用户只需运行简单的命令,而不需要手动整合和解释来自多个工具的输出。
再比如MACS2(Model-based Analysis of ChIP-Seq 2),它是一款用于分析染色质免疫共沉淀测序(ChIP-seq)等表观ngs组学数据的生物信息学工具。主要功能是寻找ChIP-seq数据中染色质峰值的位置。这些峰值通常代表着与蛋白质结合的区域,如转录因子结合位点或组蛋白修饰的区域。使用统计模型来估计ChIP-seq信号在基因组上的分布,然后通过比较实验组与对照组来确定峰值。这使得它对于各种不同的实验设计和信号强度具有一定的灵活性。
上面的这些软件本身就是一个单一的python模块,所以只需要使用Python模块安装方式即可安装软件:
# https://multiqc.info/docs/getting_started/installation/
pip install multiqc
pip install MACS2
实际上,它们虽然说就是单一的Python模块,它也是会大量依赖于其它基础Python模块,尤其是numpy 和pandas,它们直接经常是有版本冲突。而且Python本身也有不同的划时代的版本,不过起码Python 2 和 Python 3 的基本语法和编程概念是相似的。然而,由于 Python 2 已于 2020 年停止官方支持,建议新项目使用 Python 3,而已有项目则应考虑迁移。有一些工具和指南可以帮助进行平滑迁移,例如 2to3 工具,以及官方提供的迁移指南。
看起来单个python模块架构的软件非常容易安装,就是简单的 pip install 即可,但是因为它往往是依赖于特定的Python版本和Python其它模块,而且也取决于操作系统,以至于其实往往是也很难一次性安装成功,比如华大基因出品的空间单细胞转录组数据处理软件stereopy,大家可以试试看,在自己的机器上面安装它:
# https://stereopy.readthedocs.io/en/latest/content/00_Installation.html
pip install stereopy
还有肿瘤拷贝数变异的软件cnvkit虽然说也是Python的单个模块,其实往往是需要借助conda来管理。
多个模块多个命令
有一些开发者会把能实现一系列生物信息学数据分析需求的软件命令集成在同一个软件里面,比如deepTools就是如此, 在 https://deeptools.readthedocs.io/en/latest/content/list_of_tools.html 可以看到它里面的软件命令列表就非常复杂:
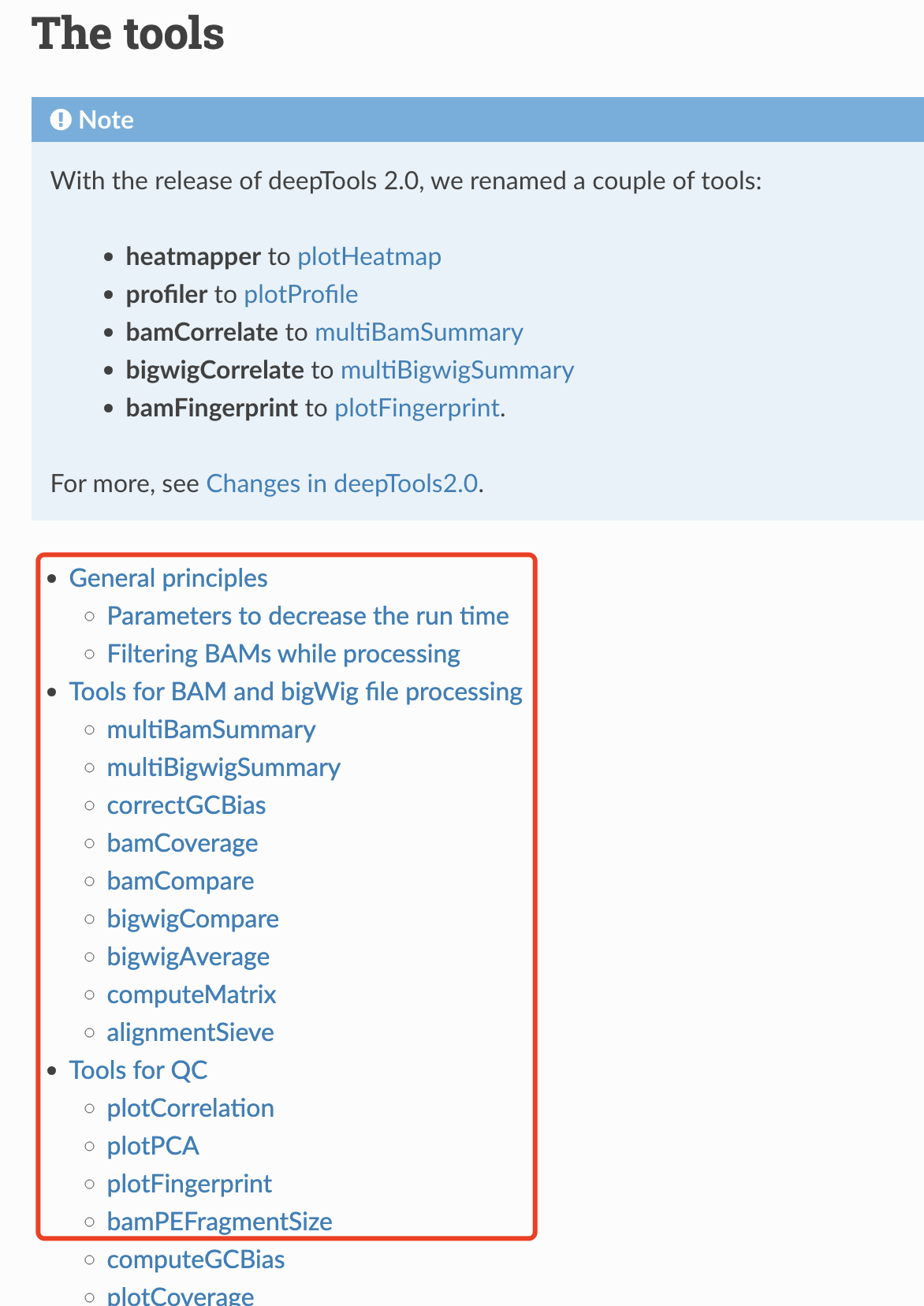
但是它的安装方法仍然是普通的简单的 pip install 即可,如下所示的命令:
pip install deeptools
同样的,它也是依赖于特定的Python版本和Python其它模块,而且也取决于操作系统,就算是你一次性安装成功后,如果Python版本或者其依赖的模块版本不准确仍然是在调用它的那些软件命令子集时候会报错哦。
基于R编程语言的软件
R编程语言跟前面的Perl和Python比起来算是后起之秀,而且它并不是默认安装在常见的操作系统里面的,无论是个人电脑的Windows还是MacOS系统,还是服务器级别的Ubuntu或者centos,理论上需要先安装R编程语言软件本身,然后才能是安装使用R编程语言制作好的软件。
其实R编程语言的软件也跟前面的Python类似,很难区分软件和R包的界限,也是有一些软件是单个R包而有一些软件需要大量的R包。不过呢,纯粹基于R编程语言的生物信息学软件其实并不多,目前主流的R功能单元仍然是脚本文件或者R包。
其它编程语言的软件
MATLAB开发的软件,比如GISTIC2软件等
ruby开发的软件,比如: http://bioruby.org/
Julia开发的软件,比如:https://github.com/BioJulia
go开发的软件,比如:https://github.com/biogo/biogo
混合多种编程语言的软件
比如转录组数据的可变剪切工具:LeafCutter software, https://github.com/davidaknowles/leafcutter;
- 第一步: bam2junc 是一个 shell脚本 (bam2junc.sh)
- 第二步:Intron clustering 是一个Python脚本 (leafcutter_cluster.py )
- 第三步:制作分组矩阵进行差异分析,是一个R脚本(leafcutter_ds.R )
- 第四步:可视化那些找到的可变剪切,是一个R脚本(ds_plots.R )
二进制可执行程序
几乎所有的编程语言开发的软件,都是可以有二进制可执行程序版本,前提是软件开发工作者愿意花费时间去制作,因为各个软件都需要在不同操作系统上面测试,最后提供稳定的二进制可执行程序下载后解压即可直接使用。
比如:https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/current/
decryption.current-centos_linux64.tar.gz 2016-07-14 15:36 6.0M
decryption.current-mac32.tar.gz 2015-02-23 17:45 5.1M
decryption.current-mac64.tar.gz 2016-07-14 15:37 5.4M
decryption.current-ubuntu32.tar.gz 2015-02-23 17:45 5.8M
decryption.current-ubuntu64.tar.gz 2016-07-14 15:37 6.0M
decryption.current-win32.zip 2016-07-14 15:37 2.6M
decryption.current-win64.zip 2016-07-14 15:37 2.9M
hisat2-2.2.1-linux.zip 2023-08-21 22:55 22M
hisat2-2.2.1-mac.zip 2022-02-04 16:46 8.9M
md5sum.txt 2023-08-29 12:45 1.2K
setup-apt.sh 2020-01-23 12:20 526
setup-yum.sh 2020-01-23 12:21 503
sratoolkit.current-centos_linux64-cloud-dbg.tar.gz 2023-08-21 22:50 176M
sratoolkit.current-centos_linux64-cloud.tar.gz 2023-08-21 22:49 89M
sratoolkit.current-centos_linux64.tar.gz 2023-08-21 22:49 89M
sratoolkit.current-mac64.tar.gz 2023-08-21 22:49 85M
sratoolkit.current-ubuntu64.tar.gz 2023-08-21 22:49 89M
sratoolkit.current-win64.zip 2023-08-21 22:49 46M
sratoolkit.current.version 2023-08-21 22:50 6
以及 ncbi-blast 的二进制可执行程序软件列表 :
ls -lh |cut -d" " -f 7-
21M 10 19 21:01 ncbi-blast-2.15.0+-3.src.rpm
63B 10 19 21:06 ncbi-blast-2.15.0+-3.src.rpm.md5
202M 10 19 21:01 ncbi-blast-2.15.0+-3.x86_64.rpm
66B 10 19 21:06 ncbi-blast-2.15.0+-3.x86_64.rpm.md5
224M 10 20 16:48 ncbi-blast-2.15.0+-aarch64-linux.tar.gz
74B 10 20 16:48 ncbi-blast-2.15.0+-aarch64-linux.tar.gz.md5
27M 10 19 21:06 ncbi-blast-2.15.0+-src.tar.gz
64B 10 19 21:06 ncbi-blast-2.15.0+-src.tar.gz.md5
31M 10 19 21:06 ncbi-blast-2.15.0+-src.zip
61B 10 19 21:06 ncbi-blast-2.15.0+-src.zip.md5
129M 10 19 21:00 ncbi-blast-2.15.0+-win64.exe
63B 10 19 21:06 ncbi-blast-2.15.0+-win64.exe.md5
245M 10 19 21:04 ncbi-blast-2.15.0+-x64-linux.tar.gz
70B 10 19 21:06 ncbi-blast-2.15.0+-x64-linux.tar.gz.md5
205M 10 19 21:06 ncbi-blast-2.15.0+-x64-macosx.tar.gz
71B 10 19 21:06 ncbi-blast-2.15.0+-x64-macosx.tar.gz.md5
133M 10 19 21:01 ncbi-blast-2.15.0+-x64-win64.tar.gz
70B 10 19 21:06 ncbi-blast-2.15.0+-x64-win64.tar.gz.md5
208M 10 19 21:04 ncbi-blast-2.15.0+.dmg
57B 10 19 21:06 ncbi-blast-2.15.0+.dmg.md5
只需要按照自己的操作系统版本下载对应的压缩包文件解压后即可
有图形用户界面(GUI)的软件
通常情况下,生物信息学软件并不会开发图形用户界面(GUI),这一现象有几个原因:
- 灵活性和自动化:生物信息学通常涉及大规模的数据处理和分析。命令行界面提供了更多的灵活性和自动化选项,使得用户可以轻松地将多个软件工具串联起来,形成复杂的分析流程。通过脚本编写,研究人员可以更好地管理、重复和分享分析过程。
- 跨平台性:命令行界面在不同操作系统上的使用更为一致。由于生物信息学研究人员可能使用不同类型的操作系统(如Linux、macOS、Windows),采用命令行界面可以使软件更具有跨平台性。
- 资源消耗:一些生物信息学任务需要大量的计算资源,命令行界面通常更有效地利用系统资源。在服务器集群上运行命令行任务比在图形界面上运行更为常见。
- 程序员导向:生物信息学领域的用户中有很多具有编程经验的研究人员。对于这些人来说,命令行界面提供了更直观和熟悉的方式来执行任务。
- 数据处理效率:命令行界面在处理大规模数据时通常更高效。对于一些需要处理数百GB或TB级别数据的任务,通过脚本编写和命令行界面执行可以提高效率。
而且,如果生物信息学软件有了图形用户界面(GUI),其实就类似于网页工具了,本地版的,可以使得非专业的生物学研究人员更容易上手和使用这些工具,降低使用门槛。GUI通常更适合初学者或非专业程序员,而命令行界面则更适合有编程经验的研究人员。
比如Cytoscape ,它是一款网络可视化工具,主要用于可视化和分析生物学网络。它能够绘制和分析分子互作网络、信号传导通路等,同时支持网络的进化和动态变化的可视化。
还有 IGV(Integrative Genomics Viewer),甚至fastqc软件本身也可以是图形界面使用它。
conda软件管理方案
也是有非常多的细节,等后面一起整理吧:
- conda下载
- miniconda安装
- miniconda配置镜像
- 创建小环境
- 查看小环境
- 进入小环境
- 查找软件
- 安装软件
- 指定软件安装版本
- 更新软件
- 查看已安装软件
- 退出小环境
- 移除小环境
不同操作系统的软件管理仓库
比如前面的网页工具开发,通常是会涉及到MySQL,以及PHP或者JavaScript,这些往往是需要管理员权限,如果是个人电脑Windows或者MacOS其实蛮简单的,都是有界面段的软件管理仓库,鼠标点击任意软件都可以自动下载。其实这个也应该是我们生物信息学软件的理想下载模式,目前借助于conda我们勉强能实现在服务器上面使用单一命令任意安装绝大部分生物信息学软件。不过,如果是有服务器的管理员权限,其实也可以借助于软件管理仓库,比如在Ubuntu操作系统来安装必备的库文件以及软件,代码如下:
sudo apt update
# sudo apt install -y net-tools
# sudo apt install -y openssh-server
sudo apt install -y vim tree nginx htop cmake
sudo apt install --fix-missing libcurl4-openssl-dev libxml2-dev libgdal-dev libssl-dev libglu1-mesa-dev libmagick++-dev libudunits2-dev
sudo apt install -y subversion scons libfuse-dev gcc git make
# sudo apt install -y samtools bcftools bwa ncbi-blast+ sra-toolkit
上面的 apt install之所以可以安装任意生物信息学软件,比如 samtools bcftools bwa ncbi-blast+ sra-toolkit , 就是因为它们加入了操作系统的软件管理仓库。但是也有一些软件是需要自己去官网下载的,比如安装一些R相关网页服务(shiny和Rstudio),因为网络问题,直接从其它腾讯云服务器拷贝两个deb文件过来。
- https://rstudio.com/products/rstudio/download-server/debian-ubuntu/
- https://rstudio.com/products/shiny/download-server/ubuntu/
如果是网络问题需要等待,有了deb文件就很容易使用gdebi命令在Ubuntu操作系统下面安装软件啦,具体的代码如下所示:
sudo apt install gdebi-core
# Install for Debian 10 / Ubuntu 18 / Ubuntu 20
wget https://download2.rstudio.org/server/bionic/amd64/rstudio-server-1.4.1103-amd64.deb
sudo gdebi rstudio-server-1.4.1103-amd64.deb
sudo su - \
-c "R -e \"install.packages('shiny', repos='https://cran.rstudio.com/')\""
# Download Shiny Server for Ubuntu 16.04 or later
wget https://download3.rstudio.org/ubuntu-14.04/x86_64/shiny-server-1.5.16.958-amd64.deb
sudo gdebi shiny-server-1.5.16.958-amd64.deb