本笔记会被收录于《生信技能树》公众号的《单细胞2024》专辑,而且我们从2024开始的教程都是基于Seurat的V5版本啦,之前已经演示了如何读取不同格式的单细胞转录组数据文件,如下所示:
因为这个Seurat的V5版本还是有一些优势的,比如可以轻轻松松拿捏这130万单细胞的数据集,需要参考Seurat官网的3个资料:
-
https://satijalab.org/seurat/articles/seurat5_sketch_analysis
-
https://satijalab.org/seurat/articles/seurat5_bpcells_interaction_vignette
就是BPCells这个R包可以通过on-disk storage的方法来读取和存储130万单细胞的数据集,然后Sketching这个方法可以从130万单细胞的数据集里面抽样但是还保留数据集的特性。
安装BPCells包
首先是安装和加载必备的包:
remotes::install_github("bnprks/BPCells")
# https://api.github.com/repos/bnprks/BPCells/tarball/HEAD
library(BPCells)
library(Seurat)
library(SeuratObject)
library(SeuratDisk)
library(Azimuth)
# needs to be set for large dataset analysis
options(future.globals.maxSize = 1e9)
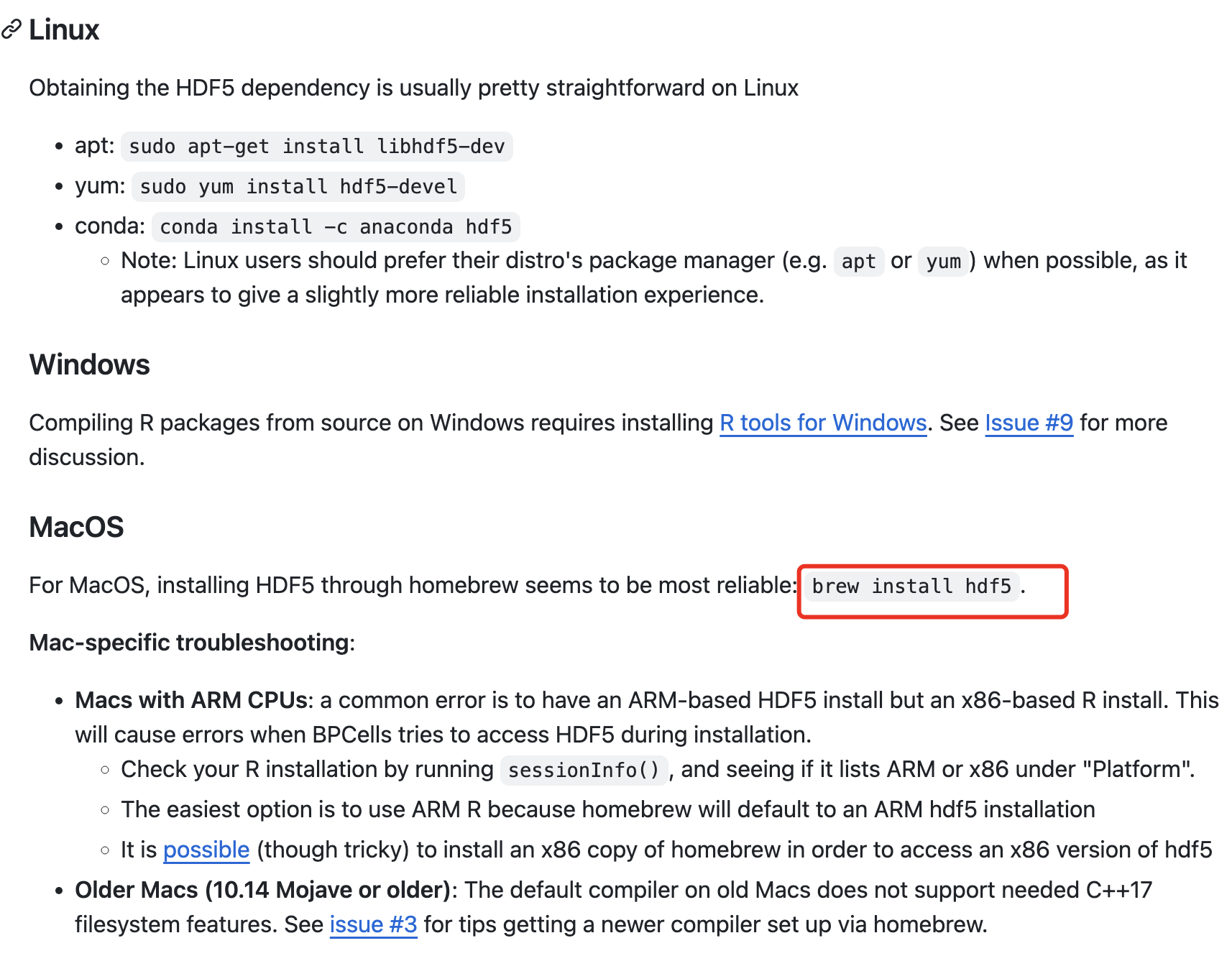
因为 BPCells 包目前仍然是在github存放,所以网络首先是一个拦路虎。然后如果你使用的并不是Windows操作系统你大概率会缺hdf5;
Searching for hdf5 in a conda env...
Unable to locate libhdf5. Please install manually or edit compiler flags.
ERROR: configuration failed for package ‘BPCells’
不同的操作系统有对应的解决方案,就是安装hdf5即可 :
全部搞定了之后就可以轻轻松松加载和使用BPCells这个R包。
查看和读取130万单细胞的数据集(h5文件)
案例的130万单细胞的数据集是10x公司在其官网提供的,链接是: https://support.10xgenomics.com/single-cell-gene-expression/datasets/1.3.0/1M_neurons?
需要简单的登陆即可看到下载链接哦,可以看到仅仅是表达量矩阵文件就有接近4G大小拉: https://cf.10xgenomics.com/samples/cell-exp/1.3.0/1M_neurons/1M_neurons_filtered_gene_bc_matrices_h5.h5
接下来让我们看看BPCells这个R包是如何操作这个h5文件吧,
brain.data <- open_matrix_10x_hdf5(
path = "1M_neurons_filtered_gene_bc_matrices_h5.h5"
)
# Write the matrix to a directory
write_matrix_dir(
mat = brain.data,
dir = 'brain_counts')
# Now that we have the matrix on disk, we can load it
brain.mat <- open_matrix_dir(dir = "brain_counts")
brain.mat <- Azimuth:::ConvertEnsembleToSymbol(mat = brain.mat, species = "mouse")
# Create Seurat Object
brain <- CreateSeuratObject(counts = brain.mat)
可以看到主要是3个步骤就把h5文件里面的单细胞转录组表达量矩阵读入到R里面成为了稀疏矩阵,简单的进行基因id转换后就可以在Seurat里面创建 Seurat 对象。下面是对每个步骤的解释:
open_matrix_10x_hdf5: 从一个 10x Genomics 的 HDF5 文件中读取单细胞转录组数据。这个数据通常包含了单细胞测序的原始计数信息。write_matrix_dir: 将读取的单细胞转录组数据写入指定的目录。这一步的目的可能是将数据存储在磁盘上,以便后续的分析。open_matrix_dir: 从指定目录中读取单细胞转录组数据。这个步骤是为了确保数据已经被写入磁盘,并且可以方便地进行后续的操作。Azimuth:::ConvertEnsembleToSymbol: 使用 Azimuth 包中的函数,将基因的 Ensembl 标识转换为符号。Ensembl 是一种基因的标识方法,转换为符号可能更容易理解和使用。CreateSeuratObject: 使用 Seurat 包中的函数,基于给定的转录组数据创建一个 Seurat 对象。Seurat 是一个用于单细胞转录组分析的流行 R 包。
整个流程的目的是将原始的单细胞转录组数据读取、存储、转换,并最终创建一个 Seurat 对象,以便进行后续的单细胞分析。比如可以简单的看看一些基因的表达量情况:
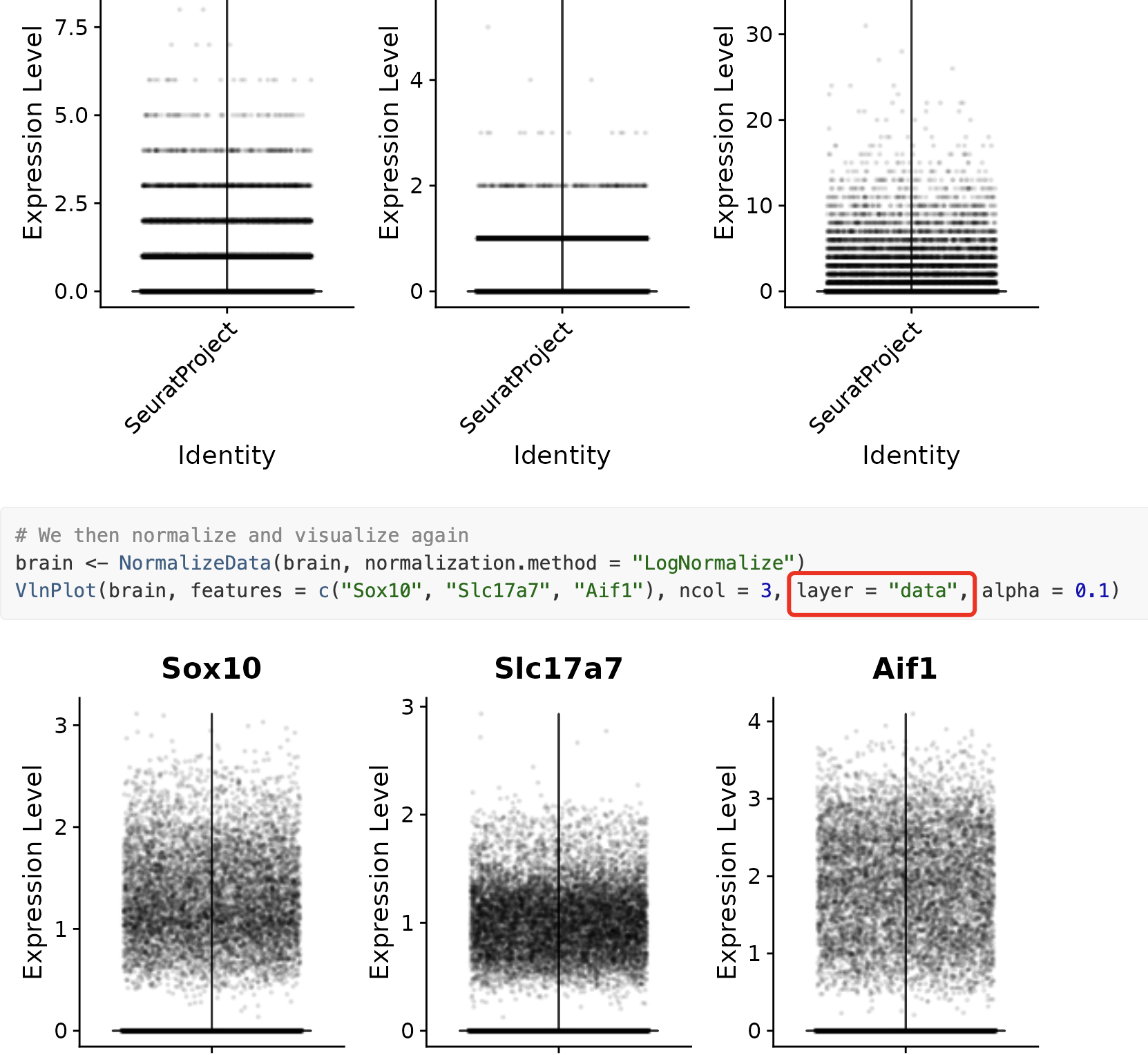
VlnPlot(brain, features = c("Sox10", "Slc17a7", "Aif1"), ncol = 3,
layer = "counts", alpha = 0.1)
# We then normalize and visualize again
brain <- NormalizeData(brain, normalization.method = "LogNormalize")
VlnPlot(brain, features = c("Sox10", "Slc17a7", "Aif1"), ncol = 3,
layer = "data", alpha = 0.1)
最开始的Seurat对象里面仅仅是有counts矩阵(纯粹的整数), 但是NormalizeData函数处理后就有了data的信息,前后可视化就可以看到区别:
这个Seurat对象也可以常规保存为R语言里面的rds文件,如下所示:
SaveSeuratRds(
object = brain,
file = "1p3_million_mouse_brain.rds"
)
后面就可以直接使用 readRDS 读取保存好的R语言里面的rds文件,而不需要从h5格式的单细胞表达量矩阵文件开始啦。
抽样走下游降维聚类分群
虽然前面我们借助了BPCells这个R包把h5文件里面的单细胞转录组表达量矩阵读入到R里面了而且创建了Seurat对象,但是直接对这个Seurat对象走下游降维聚类分群仍然是每个步骤都消耗的资源很可怕。这个时候还需要借助Sketching这个方法可以从130万单细胞的数据集里面抽样但是还保留数据集的特性,首先读取前面保存好的R语言里面的rds文件:
# Read the Seurat object, which contains 1.3M cells stored on-disk as part of the 'RNA' assay
obj <- readRDS("1p3_million_mouse_brain.rds")
obj
## An object of class Seurat
## 27282 features across 1306127 samples within 1 assay
## Active assay: RNA (27282 features, 0 variable features)
## 1 layer present: counts
# Note that since the data is stored on-disk, the object size easily fits in-memory (<1GB)
format(object.size(obj), units = "Mb")
## [1] "596.2 Mb"
然后抽样:
obj <- NormalizeData(obj)
obj <- FindVariableFeatures(obj)
obj <- SketchData(
object = obj,
ncells = 50000,
method = "LeverageScore",
sketched.assay = "sketch"
)
obj
接下来就降维聚类分群即可;
DefaultAssay(obj) <- "sketch"
obj <- FindVariableFeatures(obj)
obj <- ScaleData(obj)
obj <- RunPCA(obj)
obj <- FindNeighbors(obj, dims = 1:50)
obj <- FindClusters(obj, resolution = 2)
obj <- RunUMAP(obj, dims = 1:50, return.model = T)
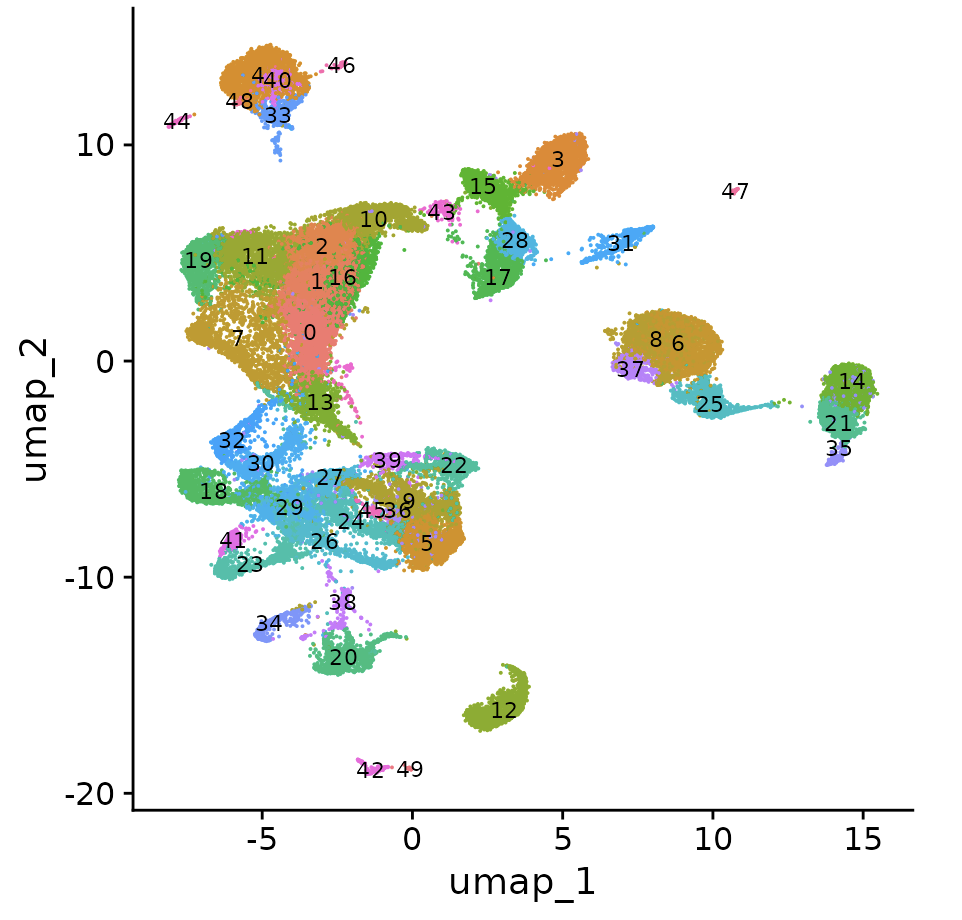
DimPlot(obj, label = T, label.size = 3, reduction = "umap") + NoLegend()
可以看到,虽然说是抽样后的子集,其实细胞数量也很可观的:
因为是小鼠大脑,所以可以根据生物学背景知识简单的看一下常见的各个单细胞亚群特异性高表达量基因的情况:
FeaturePlot(
object = obj,
features = c(
"Igfbp7", "Neurod6", "Dlx2", "Gad2",
"Eomes", "Vim", "Reln", "Olig1", "C1qa"
),
ncol = 3
)
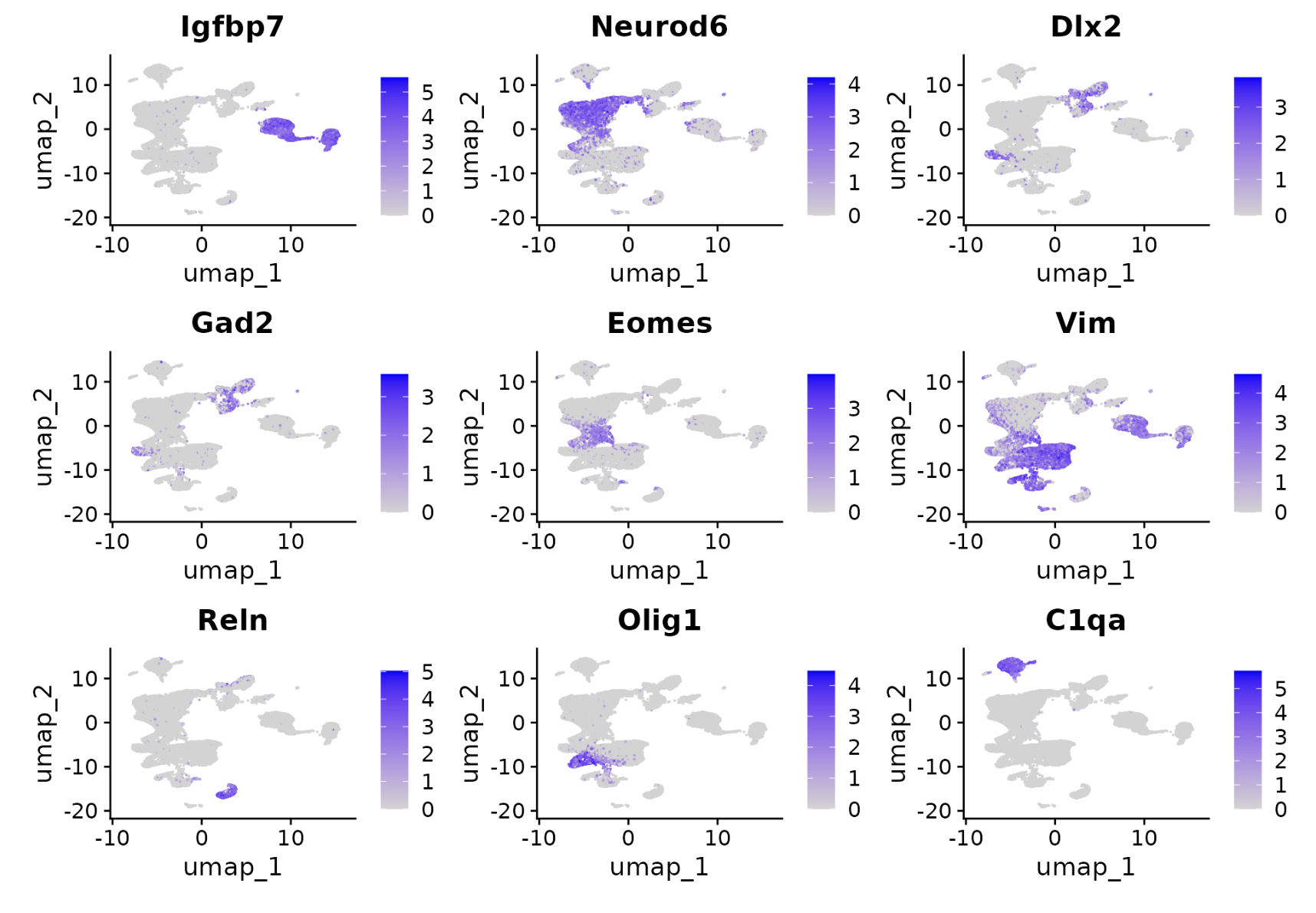
在子集的降维聚类分群结果,可以映射到前面的完整的数据集,就是130万单细胞,这个时候使用ProjectData函数即可:
obj <- ProjectData(
object = obj,
assay = "RNA",
full.reduction = "pca.full",
sketched.assay = "sketch",
sketched.reduction = "pca",
umap.model = "umap",
dims = 1:50,
refdata = list(cluster_full = "seurat_clusters")
)
# now that we have projected the full dataset, switch back to analyzing all cells
DefaultAssay(obj) <- "RNA"
DimPlot(obj, label = T, label.size = 3, reduction = "full.umap", group.by = "cluster_full", alpha = 0.1) + NoLegend()
可以看到,其实是差不多的umap图形,区别就是细胞数量多了很多:
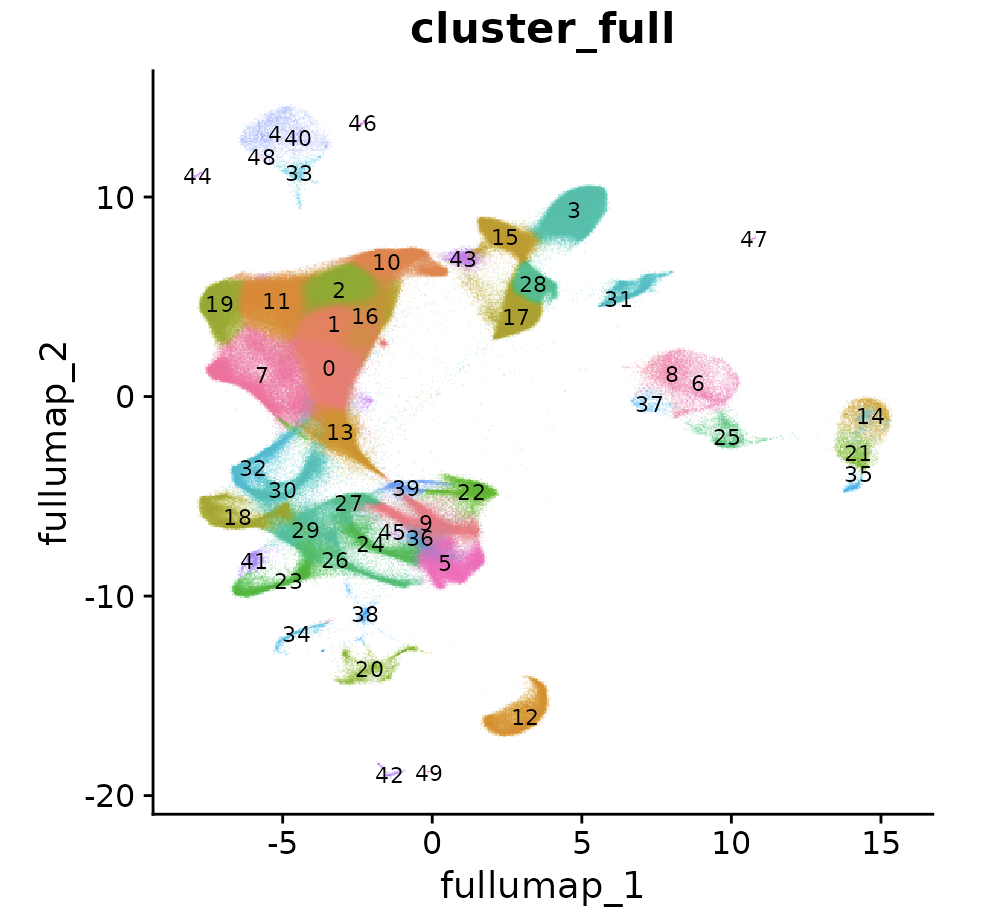
值得注意的是,这个时候我们的Seurat对象里面有两个不同的Assay,需要仔细区分哦:
# visualize gene expression on the sketched cells (fast) and the full dataset (slower)
# switch to analyzing the sketched dataset (in-memory)
DefaultAssay(obj) <- "sketch"
x1 <- FeaturePlot(obj, "C1qa")
# switch to analyzing the full dataset (on-disk)
DefaultAssay(obj) <- "RNA"
x2 <- FeaturePlot(obj, "C1qa")
x1 | x2
我们之前提到的Seurat的5种常见的可视化都是可以在这两个不同的Assay上面测试的哦。
取子集后就无需抽样了
前面的数据集是130万单细胞,但是降维聚类分群后,如果我们仅仅是感兴趣里面的部分亚群,比如从上面的UMAP里面挑选出来 2, 15, 18, 28, 40这些亚群,其实就20万左右的细胞数量了,可以无需抽样,直接让它成为存储了稀疏矩阵在内存里面的正常的Seurat对象,然后常规的走降维聚类分群即可;
# subset cells in these clusters. Note that the data remains on-disk after subsetting
obj.sub <- subset(obj, subset = cluster_full %in% c(2, 15, 18, 28, 40))
DefaultAssay(obj.sub) <- "RNA"
# now convert the RNA assay (previously on-disk) into an in-memory representation (sparse Matrix)
# we only convert the data layer, and keep the counts on-disk
obj.sub[["RNA"]]$data <- as(obj.sub[["RNA"]]$data, Class = "dgCMatrix")
# recluster the cells
obj.sub <- FindVariableFeatures(obj.sub)
obj.sub <- ScaleData(obj.sub)
obj.sub <- RunPCA(obj.sub)
obj.sub <- RunUMAP(obj.sub, dims = 1:30)
obj.sub <- FindNeighbors(obj.sub, dims = 1:30)
obj.sub <- FindClusters(obj.sub)
DimPlot(obj.sub, label = T, label.size = 3) + NoLegend()
20万左右的细胞数量是我们的普通电脑或者服务器是可以hold住的啦, 大家啊可以试试看我们之前收集整理的大脑区域的不同单细胞亚群的特异性基因,看看在这个数据集里面是否有效。详见 :两个神经退行性疾病的单细胞核转录组队列的细胞亚群及其标记基因的比较,我列出来了一些基因:
astrocytes = c("AQP4", "ADGRV1", "GPC5", "RYR3")
endothelial = c("CLDN5", "ABCB1", "EBF1")
excitatory = c("CAMK2A", "CBLN2", "LDB2")
inhibitory = c("GAD1", "LHFPL3", "PCDH15")
microglia = c("C3", "LRMDA", "DOCK8")
oligodendrocytes = c("MBP", "PLP1", "ST18")
OPC='Tnr,Igsf21,Neu4,Gpr17'
Ependymal='Cfap126,Fam183b,Tmem212,pifo,Tekt1,Dnah12'
pericyte=c( 'DCN', 'LUM', 'GSN' ,'FGF7','MME', 'ACTA2','RGS5')
# 下面是4种神经细胞
# excitatory (SLC17A6),
# inhibitory (GAD2),
# GABAergic (GAD2/GRIK1),
# dopaminergic neurons(TH)
其中 neurons 还可以细分为:
- motoneurons (MN),
- excitatory dorsal neurons (ExDorsal),
- inhibitory dorsal neurons (InhDorsal),
- excitatory mid and ventral neurons (ExMV),
- inhibitory or mixed mid and ventral neurons (InhMV).
也可以更细,取决于各自的生物学背景啦。