前些天在《生信技能树》的微信视频号做了一个肿瘤单细胞转录组的数据分析直播,文章是:《Delineating the dynamic evolution from preneoplasia to invasive lung adenocarcinoma by integrating single-cell RNA sequencing and spatial transcriptomics》详见:换一个分析策略会导致文章的全部论点都得推倒重来吗。
主要的分析就是第一层次降维聚类分群,然后大概认识一下有什么亚群,以及比例差异情况,最后就是把每个亚群都细分一下做同样的分析即可。
认识GEO数据库里面的单细胞转录组数据文件格式
我们《生信菜鸟团》的单细胞周更专辑作者分享过好几次了基础文件读取技巧啦,详见: 读取不同格式的单细胞转录组数据及遇到问题的解决办法。其中最常见的就是使用Read10X读取3个文件,但是Read10X读取3个文件还得注意版本,而且必须保证3个文件名字完全一样,要么是
barcodes.tsv.gz features.tsv.gz matrix.mtx.gz
或者说是:
barcodes.tsv genes.tsv matrix.mtx
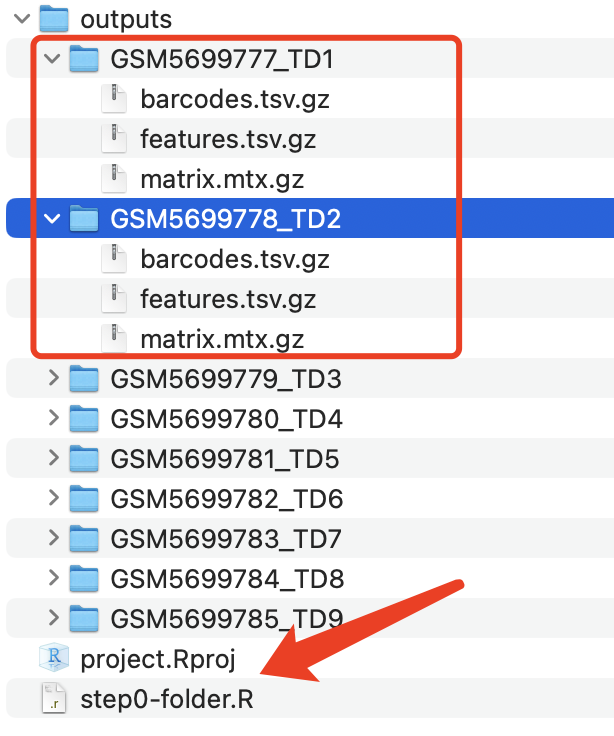
只有这样才能把表达量矩阵读入进去。而这个数据集是如下所示的文件:
GSM5699777_TD1_barcodes.tsv.gz 73.7 Kb
GSM5699777_TD1_features.tsv.gz 297.6 Kb
GSM5699777_TD1_matrix.mtx.gz 71.4 Mb
GSM5699778_TD2_barcodes.tsv.gz 86.0 Kb
GSM5699778_TD2_features.tsv.gz 297.6 Kb
GSM5699778_TD2_matrix.mtx.gz 77.3 Mb
所以是需要改名的!
整理和组织文件夹
├── [4.0K] TD1
│ ├── [ 74K] barcodes.tsv.gz
│ ├── [298K] features.tsv.gz
│ └── [ 71M] matrix.mtx.gz
├── [4.0K] TD2
│ ├── [ 86K] barcodes.tsv.gz
│ ├── [298K] features.tsv.gz
│ └── [ 77M] matrix.mtx.gz
如下所示:
构建Seurat对象
包的Read10X函数是可以读取单个样品的一个文件夹路径,但是我们是需要循环读取每个文件夹,所以是lapply这样的读取方式:
dir='GSE189357_RAW/outputs/'
samples=list.files( dir )
samples
# samples = head(samples,10)
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
sce =CreateSeuratObject(counts = Read10X(file.path(dir,pro )) ,
project = gsub('^GSM[0-9]*_','',
gsub('filtered_feature_bc_matrix','',pro) ) ,# pro, #
min.cells = 5,
min.features = 500 )
return(sce)
})
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ],
add.cell.ids = gsub('^GSM[0-9]*_','',
gsub('filtered_feature_bc_matrix','',samples)) )
这个seurat对象的结构非常复杂,值得细细品读!
质量控制:
值得注意是我们依赖于这个V4的版本的Seurat流程做出来了大量的公共数据集的单细胞转录组降维聚类分群流程,100多个公共单细胞数据集全部的处理,链接:https://pan.baidu.com/s/1MzfqW07P9ZqEA_URQ6rLbA?pwd=3heo
下面的scRNA_scripts文件夹的r代码都在上面的百度云网盘链接(https://pan.baidu.com/s/1MzfqW07P9ZqEA_URQ6rLbA?pwd=3heo)里面哦:
###### step2:QC质控 ######
dir.create("./1-QC")
setwd("./1-QC")
# 如果过滤的太狠,就需要去修改这个过滤代码
source('../scRNA_scripts/qc.R')
sce.all.filt = basic_qc(sce.all)
print(dim(sce.all))
print(dim(sce.all.filt))
setwd('../')
如果是Seurat版本问题,参考我们前面的降级方式:假如你不喜欢最新版的Seurat包的单细胞理念
整合后再降维聚类分群
单细胞的每个样品其实都是批次,原则上这样的批次是不可以矫正的,所以这个时候我们把这个步骤称作是整合。
wc -l scRNA_scripts/*
368 scRNA_scripts/check-all-markers.R
48 scRNA_scripts/harmony.R
14 scRNA_scripts/lib.R
102 scRNA_scripts/qc.R
scRNA_scripts文件夹的r代码都在上面的百度云网盘链接(https://pan.baidu.com/s/1MzfqW07P9ZqEA_URQ6rLbA?pwd=3heo)里面哈。
确定分辨率
我们默认会看看 0.1和0.8这两个分辨率的情况:
# 为了省力,我们直接看 0.1和0.8即可
table(Idents(sce.all.int))
table(sce.all.int$seurat_clusters)
table(sce.all.int$RNA_snn_res.0.1)
table(sce.all.int$RNA_snn_res.0.8)
getwd()
dir.create('check-by-0.1')
setwd('check-by-0.1')
sel.clust = "RNA_snn_res.0.1"
sce.all.int <- SetIdent(sce.all.int, value = sel.clust)
table(sce.all.int@active.ident)
source('../scRNA_scripts/check-all-markers.R')
setwd('../')
getwd()
dir.create('check-by-0.8')
setwd('check-by-0.8')
sel.clust = "RNA_snn_res.0.8"
sce.all.int <- SetIdent(sce.all.int, value = sel.clust)
table(sce.all.int@active.ident)
source('../scRNA_scripts/check-all-markers.R')
setwd('../')
getwd()
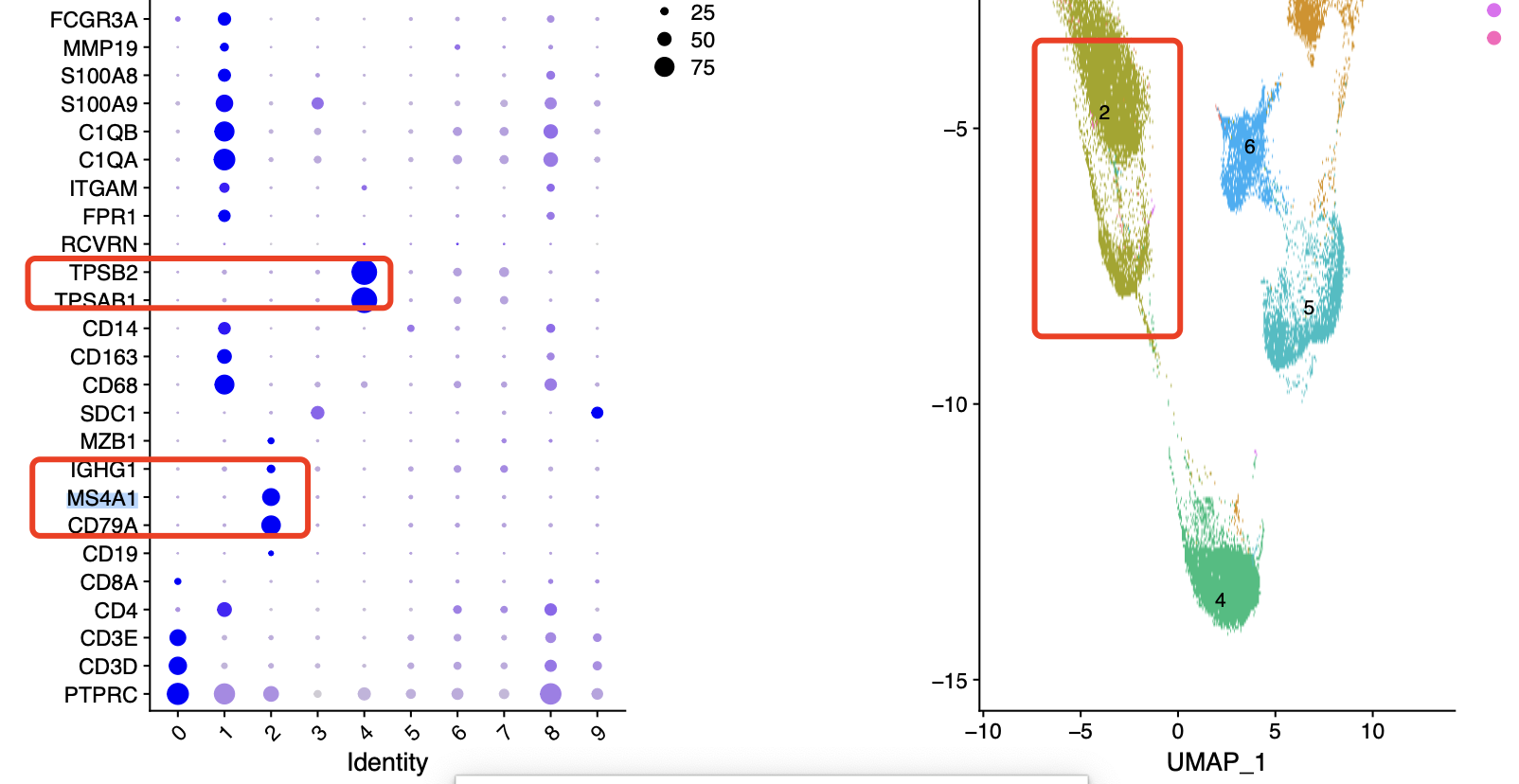
如下所示的0.1分辨率群就很少:
如下所示的0.8分辨率群就很多:
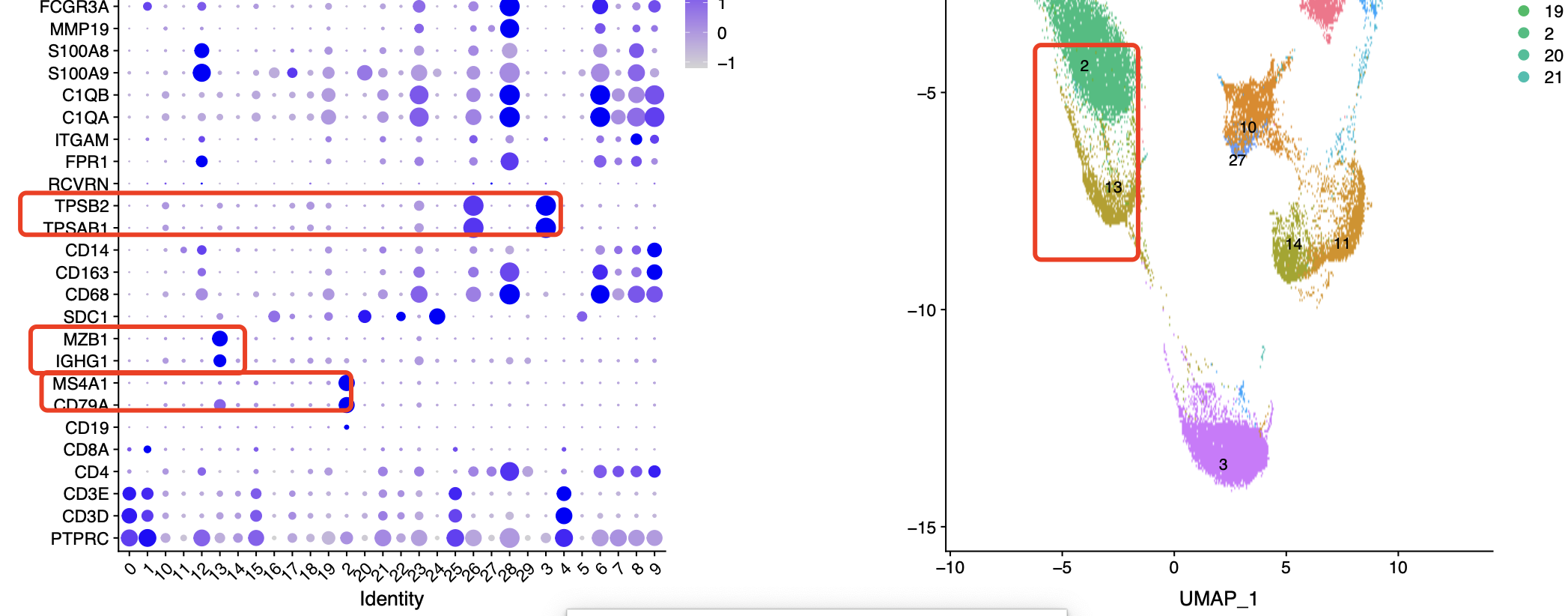
在上面的0.1的分辨率下面,b细胞和plasma细胞是没办法区分的,但是在0.8的分辨率下面可以看到2群是b细胞但是13群是plasma细胞。
人工注释单细胞亚群
肺癌单细胞数据集也有好几十个了,拿到表达量矩阵后的第一层次降维聚类分群通常是:
- immune (CD45+,PTPRC),
- epithelial/cancer (EpCAM+,EPCAM),
- stromal (CD10+,MME,fibo or CD31+,PECAM1,endo)
参考我前面介绍过 CNS图表复现08—肿瘤单细胞数据第一次分群通用规则,这3大单细胞亚群构成了肿瘤免疫微环境的复杂。所以是很容易降维聚类分群啦,我们都是人工命名,比如上面的0.1的分辨率下面就可以人工判断给出来下面的亚群名字:
#定义细胞亚群
celltype[celltype$ClusterID %in% c( 0 ),2]='Tcells'
celltype[celltype$ClusterID %in% c( 2 ),2]='Bcells'
celltype[celltype$ClusterID %in% c( 1 ),2]= 'myeloids' #
celltype[celltype$ClusterID %in% c( 4 ),2]='mast'
celltype[celltype$ClusterID %in% c( 5 ),2]='endo'
celltype[celltype$ClusterID %in% c(6),2]='fibro'
celltype[celltype$ClusterID %in% c(8),2]='cycle'
celltype[celltype$ClusterID %in% c( 3,7,9 ),2]='epi'
整体来说这个复现的代码在百度云分享给大家:链接:https://pan.baidu.com/s/1niFqyAiUU3yXK1W26b8RvQ?pwd=nbmj
如果是其它数据集
重点看数据文件如何读入,然后看细胞亚群如何人工确定名字!我们依赖于这个V4的版本的Seurat流程做出来了大量的公共数据集的单细胞转录组降维聚类分群流程,100多个公共单细胞数据集全部的处理,链接:https://pan.baidu.com/s/1MzfqW07P9ZqEA_URQ6rLbA?pwd=3heo
上面的代码演示都是在《生信技能树》的微信视频号,应该是很容易关注后看里面的回放哦!