前面我们分享了:时间序列转录组多次差异分析以及时序分析,这里面的开头是三分组的转录组测序数据,但是后面代码演示的时候是不同时间点处理的肿瘤细胞系表达量芯片数据。
因为代码是收费的,所以需要简单的回复一下读者的提问,就是大家感兴趣这个代码到底该如何移植到转录组测序数据分析,而且读者给出来了一个案例,就是2020的文章《Transcriptomic profiling across the nonalcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis》,它对应的数据集是:GSE135251,在其页面可以看到是216 snap frozen liver biopsies, comprising 206 NAFLD cases with different fibrosis stages and 10 controls were studied.
关心的是:non-alcoholic fatty liver disease (NAFLD) 的疾病进展,详见:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE135251
我们首先读取GSE135251的转录组测序表达量矩阵:
需要自行下载 https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSE135251&format=file 这个压缩包文件,并且解压在 GSE135251_RAW 文件夹里面:
rm(list = ls()) ## 魔幻操作,一键清空~
options(stringsAsFactors = F)
getOption('timeout')
options(timeout=10000)
if(T){
# https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE135251
library(AnnoProbe)
library(GEOquery)
gset <- getGEO("GSE135251",getGPL = F)
gset
gset[[1]]
a=gset[[1]] #
phe=pData(a)
# dat=exprs(a) #a现在是一个对象,取a这个对象通过看说明书知道要用exprs这个函数
# dim(dat)#看一下dat这个矩阵的维度
# dat[1:4,1:4] #查看dat这个矩阵的1至4行和1至4列,逗号前为行,逗号后为列
# boxplot(dat[,1:4],las=2)
# zscore的矩阵
# 发现并不需要log了,所以注释了下面的代码
#dat=log2(dat)
pd=phe[,(ncol(phe)-4):ncol(phe)]
}
library(ggplot2)
fs=list.files('GSE135251_RAW/', full.names = T)
fs
library(data.table)
tmp = fread(fs[1],data.table = F)
head(tmp)
gid=fread(fs[1],data.table = F)[,1]
head(gid)
rawcount = do.call(cbind,
lapply(fs, function(x){
fread(x,data.table = F)[,2]
}))
rawcount[1:4,1:4]
mat = rawcount
rownames(mat) = gsub('[.][0-9]*','', gid)
# str_split(fs,'[/.]',simplify = T)[,2]
colnames(mat) = substring(basename(fs),1,10)
keep_feature <- rowSums (mat > 1) > 1
colSums(mat)/1000000
table(keep_feature)
mat <- mat[keep_feature, ]
ensembl_matrix=mat
ensembl_matrix[1:4,1:4]
colnames(ensembl_matrix)
library(AnnoProbe)
head(rownames(ensembl_matrix))
ids=annoGene(rownames(ensembl_matrix),'ENSEMBL','human')
head(ids)
ids=ids[!duplicated(ids$SYMBOL),]
ids=ids[!duplicated(ids$ENSEMBL),]
symbol_matrix= ensembl_matrix[match(ids$ENSEMBL,rownames(ensembl_matrix)),]
rownames(symbol_matrix) = ids$SYMBOL
symbol_matrix[1:4,1:4]
#通过查看说明书知道取对象a里的临床信息用pData
## 挑选一些感兴趣的临床表型。
library(stringr)
phe=pd
# 这里一定要人工干预,每个数据集项目的分组不一样
# 是主观判断,自己选择
#group_list=ifelse(grepl('Healthy',phe$title),'healthy','patient')
group_list=phe$`disease:ch1`
save(dat,group_list,phe,symbol_matrix,file = 'step1-output.Rdata')
source('step2-check.R')
# control和disease分组的样品数量差异很大
然后是7次差异分析:
首先可以看到:
> as.data.frame(table(phe$`group in paper:ch1`))
Var1 Freq
1 control 10
2 NAFL 51
3 NASH_F0-F1 34
4 NASH_F2 53
5 NASH_F3 54
6 NASH_F4 14
分组需要结合背景知识:
> table(phe$`group in paper:ch1`,phe$`fibrosis stage:ch1`)
0 1 2 3 4
control 8 1 1 0 0
NAFL 33 18 0 0 0
NASH_F0-F1 5 29 0 0 0
NASH_F2 0 0 53 0 0
NASH_F3 0 0 0 54 0
NASH_F4 0 0 0 0 14
“NASH” 是脂肪性肝病(Non-Alcoholic Steatohepatitis)的缩写,这是一种与酒精无关的脂肪肝炎,通常与代谢综合症、肥胖和糖尿病等相关。评分系统用于评估NASH疾病的严重程度和患者的风险。以下是四种常见的NASH评分系统:
- NAFLD Activity Score (NAS) / NASH分数:
- 描述:NAS是一种常用于评估NASH严重程度的评分系统,它考察肝组织切片中的三个主要特征:脂肪变性(steatosis)、细胞损伤(ballooning)、和炎症(lobular inflammation)。
- 评分范围:通常从0到8,分数越高表示NASH的严重程度越高。
- 解释:NAS分数通常用于确定NASH的严重程度,分数≥5表示NASH,分数≥3表示NAFLD。
- Fibrosis-4指数(FIB-4):
- 描述:FIB-4用于评估NAFLD患者的肝纤维化程度,而不是NASH本身的严重程度。它使用年龄、AST(天门冬氨酸转氨酶)和ALT(丙氨酸转氨酶)水平以及血小板计数来计算。
- 评分范围:通常从1到3,分数越高表示肝纤维化的风险越高。
- 解释:FIB-4指数通常用于筛查哪些NAFLD患者可能有肝纤维化,而不是评估NASH的严重程度。
- AST to Platelet Ratio Index (APRI):
- 描述:类似于FIB-4,APRI也用于评估肝纤维化程度。它使用AST和血小板计数来计算。
- 评分范围:通常从0到2,分数越高表示肝纤维化的风险越高。
- 解释:APRI指数用于估计NAFLD患者的肝纤维化风险。
- BARD评分:
- 描述:BARD评分是一种用于评估NASH患者的肝纤维化风险的系统。它使用BMI(体重指数)、AST/ALT比率和糖尿病状况来评分。
- 评分范围:通常从0到4,分数越高表示肝纤维化的风险越高。
- 解释:BARD评分通常用于识别那些患有NASH并且存在较高肝纤维化风险的患者。
前面我们获取了非常详细的样品信息,文章是进行了4+3次差异分析,如下所示:
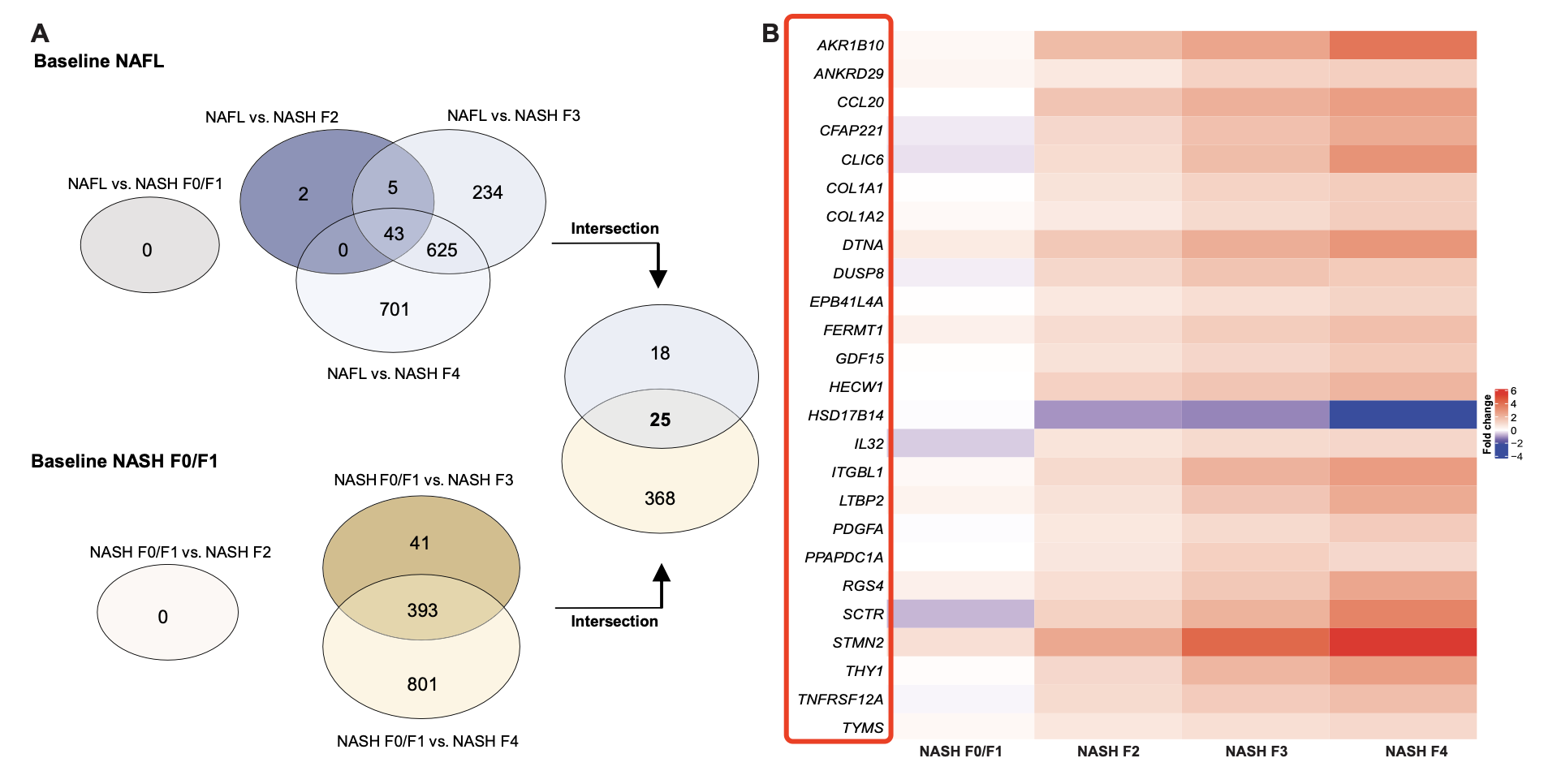
这些是可以使用我们的代码进行批量差异分析的:
rm(list = ls()) ## 魔幻操作,一键清空~
options(stringsAsFactors = F)
library(clusterProfiler)
library(stringr)
library(limma)
run <- function(){
getwd()
source('../functions/run_check.R')
run_check(dat, group_list, target_gene='GAPDH', pro=gse_number,path='./')
source('../functions/run_DEG_by_DESeq2.R')
group_list
table(group_list)
deg=run_DEG(dat, symbol_matrix,group_list, pro=gse_number,path='./',
target_gene=c('GAPDH'))
head(deg)
source('../functions/run_ORA_KEGG.R')
# Reading KEGG annotation online:
run_ORA_KEGG(deg, path='./')
source('../functions/run_ORA_GO.R')
run_ORA_GO(deg, path='./')
load(file = 'anno_DEG.Rdata')
source('../functions/run_GSEA_KEGG.R')
run_GSEA_KEGG(DEG, path='./')
}
load(file = 'step1-output.Rdata')
table(group_list)
table(phe$`fibrosis stage:ch1`)
as.data.frame(table(phe$`group in paper:ch1`))
table(phe$`group in paper:ch1`,phe$`fibrosis stage:ch1`)
# 每次都要检测数据
dat[1:4,1:4]
raw_dat = dat
raw_symbol_matrix = symbol_matrix
raw_gp = phe$`group in paper:ch1`
# NAFL NASH_F0-F1 NASH_F2 NASH_F3 NASH_F4
## 需要 写一个循环,7次差异分析
{
pro='NASH_F4-vs-NAFL'
dir.create(pro)
setwd(pro)
table(raw_gp)
kp = raw_gp %in% c('NAFL' ,'NASH_F4' )
table(kp)
dat = raw_dat[,kp]
group_list = raw_gp[kp]
symbol_matrix = raw_symbol_matrix[,kp]
gse_number = pro
save( gse_number,
dat,group_list,
file = 'step1-output.Rdata')
#load(file = 'step1-output.Rdata')
run()
setwd('../')
getwd()
}
关键分析应该是mfuzz或者wgcna
文章仅仅是根据这些样品的表达量矩阵计算了距离后进行最简单的无监督的层次聚类,然后认为的划分成为了两个亚群,但是实际上这两个亚群跟临床特征几乎是没有关联。。。。
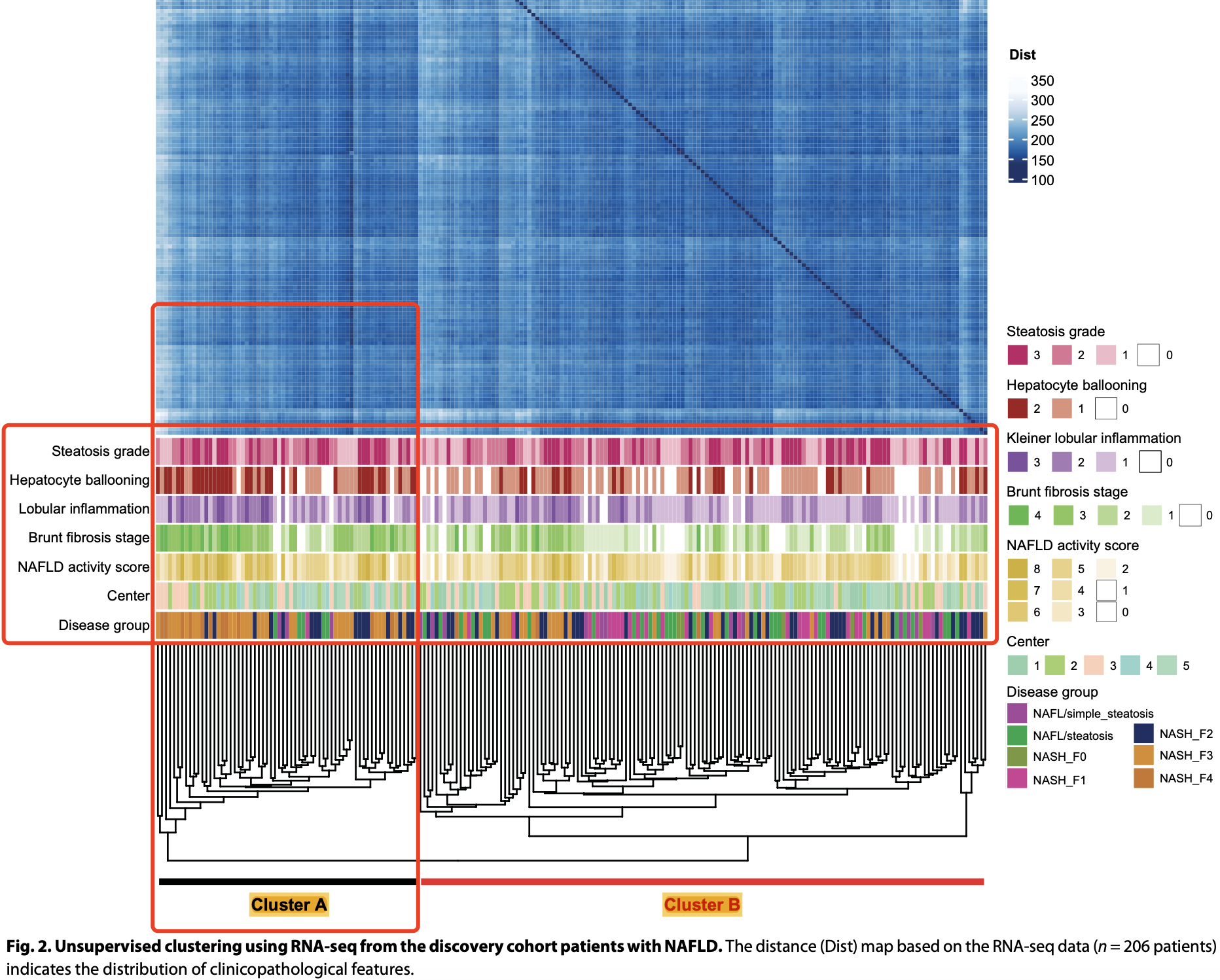
我怀疑应该是这个队列里面的最大的差异来源是取样时候的样品的细胞比例,它会最大程度影响样品本身的表达量组成,它就是无监督层次聚类的2分组的背后的决定性因素,而取样的时候的样品细胞比例状态是不可控的,所以它这个层次聚类代分组就没什么意义了。
当然了,这个仅仅是我的推测啦。即使是抛开它这个无监督层次聚类的2分组不谈,它多次差异分析取交集来定位到25个基因,这样的操作也不可取,其实mfuzz或者wgcna更好,可以参考前面我们分享的代码:时间序列转录组多次差异分析以及时序分析,比如也是默认的9个分组的基因集:
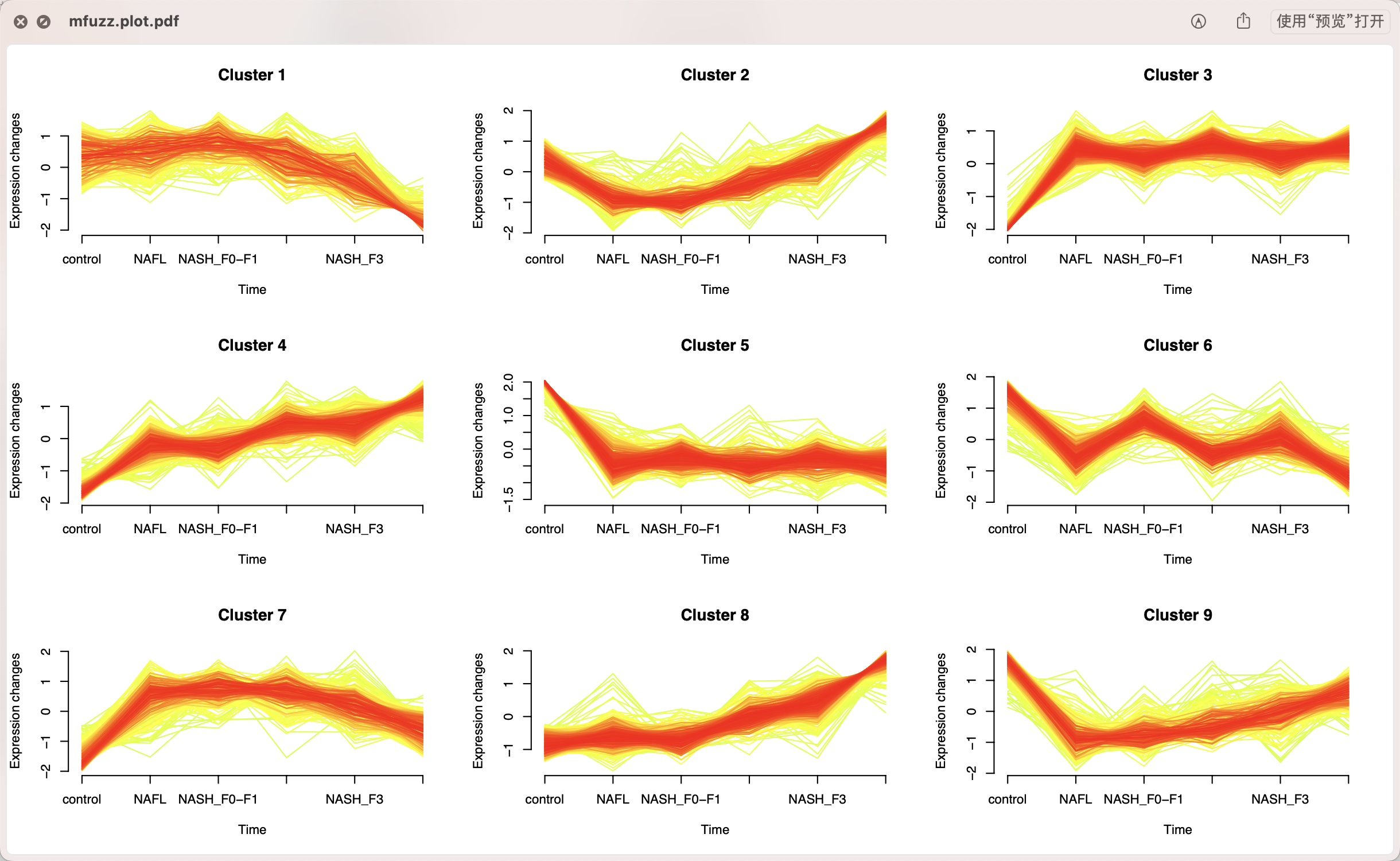
肉眼可以看到9个分组的基因集里面的,其中c1是持续下降,c4和c8是持续上升啦,但是c4和c8也有细微区别。
然后针对这9个基因集进行功能富集,因为c4和c8也有细微表达量趋势变化区别所以它们背后的功能非常类似比如都是ECM和focal adhesion但是也有差异比如ebv病毒感染。
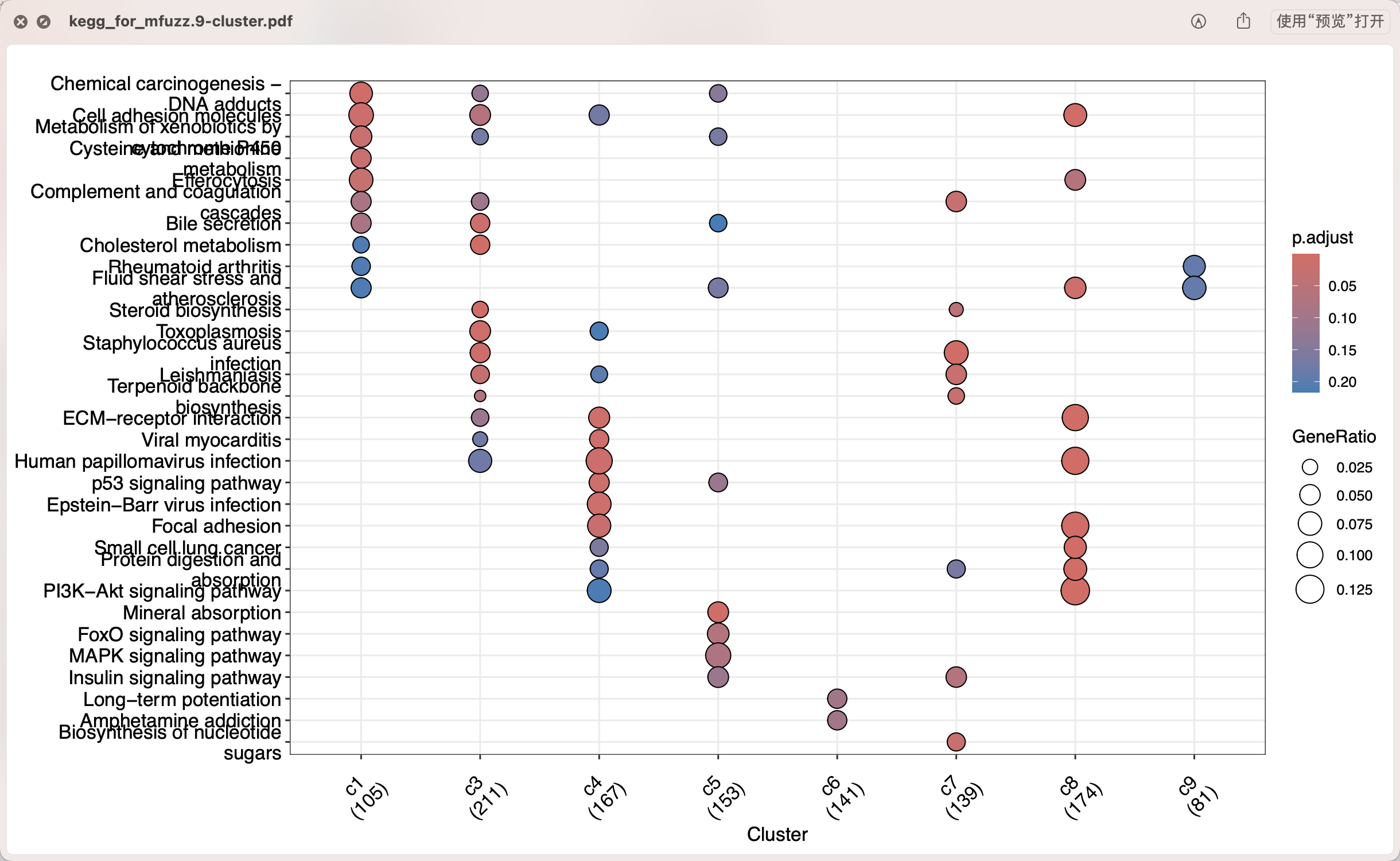
但是因为我不熟悉”NASH” 这个脂肪性肝病(Non-Alcoholic Steatohepatitis),所以没办法编造一个生物学故事啦,我虽然说可以继续wgcna分析它,但是无非是多一些图表罢了。。。。
参考前面我们分享的代码:时间序列转录组多次差异分析以及时序分析即可完成这样的图表和数据挖掘啦,超级简单!