最近蛮多粉丝在咱们《生信技能树》公众号后台咨询在肿瘤拷贝数变异数据分析,我以为说的是单细胞的inferCNV和copykat呢,因为前两天恰好分享了 比较了copykat和infercnv这两个从单细胞转录组数据推断肿瘤拷贝数变异技术差异 ,但是仔细跟粉丝在公众号后台对话才方向人家结果居然真的是咨询肿瘤外显子得到的肿瘤拷贝数变异信息。
粉丝的问题很朴素,就是想把TCGA数据库里面的非小细胞肺癌里面的肺鳞癌区分成为是否有TP53这个基因的somatic突变的两个分组,然后去比较这两个组别里面的病人的肿瘤拷贝数变异,做一个差异分析。
关于这样的分析,我们推荐一个最近发表在《NATURE GENETICS》杂志,2020的文章:《Genomic characterization of human brain metastases identifies drivers of metastatic lung adenocarcinoma》,比较了BM-LUAD 和TCGA-LUAD 两个队列的拷贝数差异情况。
首先展现两个队列拷贝数变异全景图,而且是扩增情况和缺失情况分开展现 :
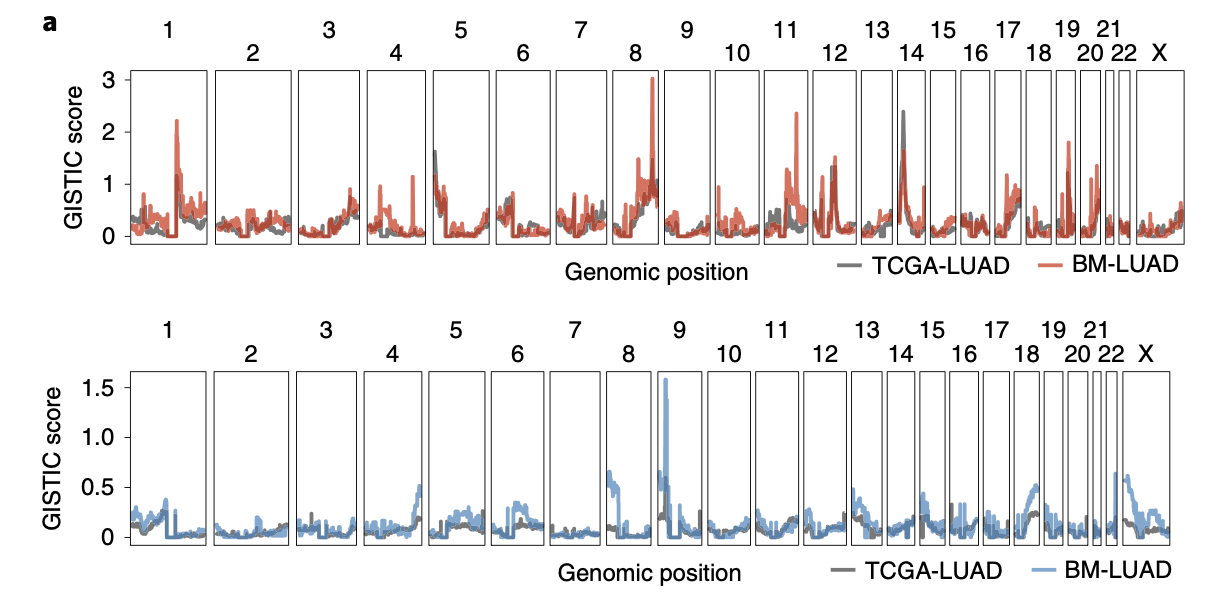
图例是: a, GISTIC amplification (top) and deletion (bottom) plots of BM-LUAD (n = 73) and covariate-matched samples in the TCGA-LUAD (n = 464) cohort
可以看到,这样的结果其实肉眼也能看到一些两个图的差异,但是毕竟没有统计学检验,这个时候两个拷贝数变异结果都可以导入GISTIC2软件,然后统计学检验后绘制散点图:
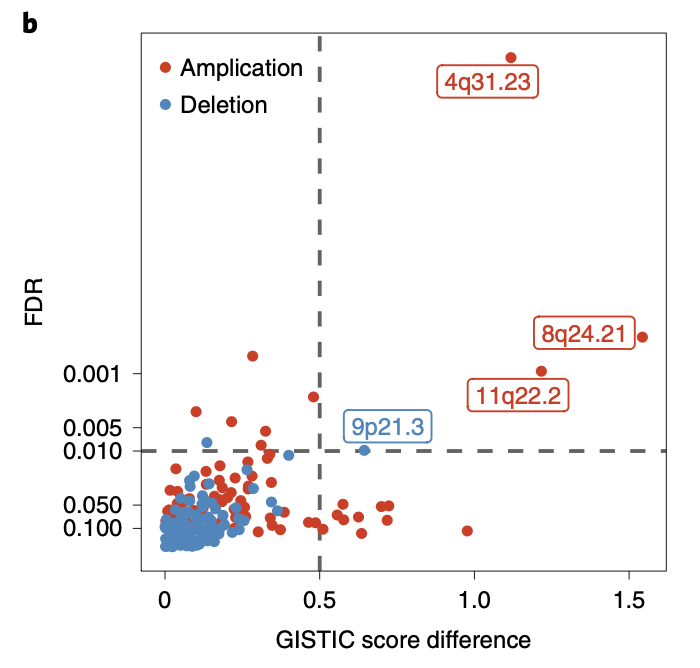
图例是:b, Differentially amplified or deleted regions in BM-LUAD compared to TCGA-LUAD. Significant differential regions are labeled (FDR < 0.01; G-score difference > 0.5).
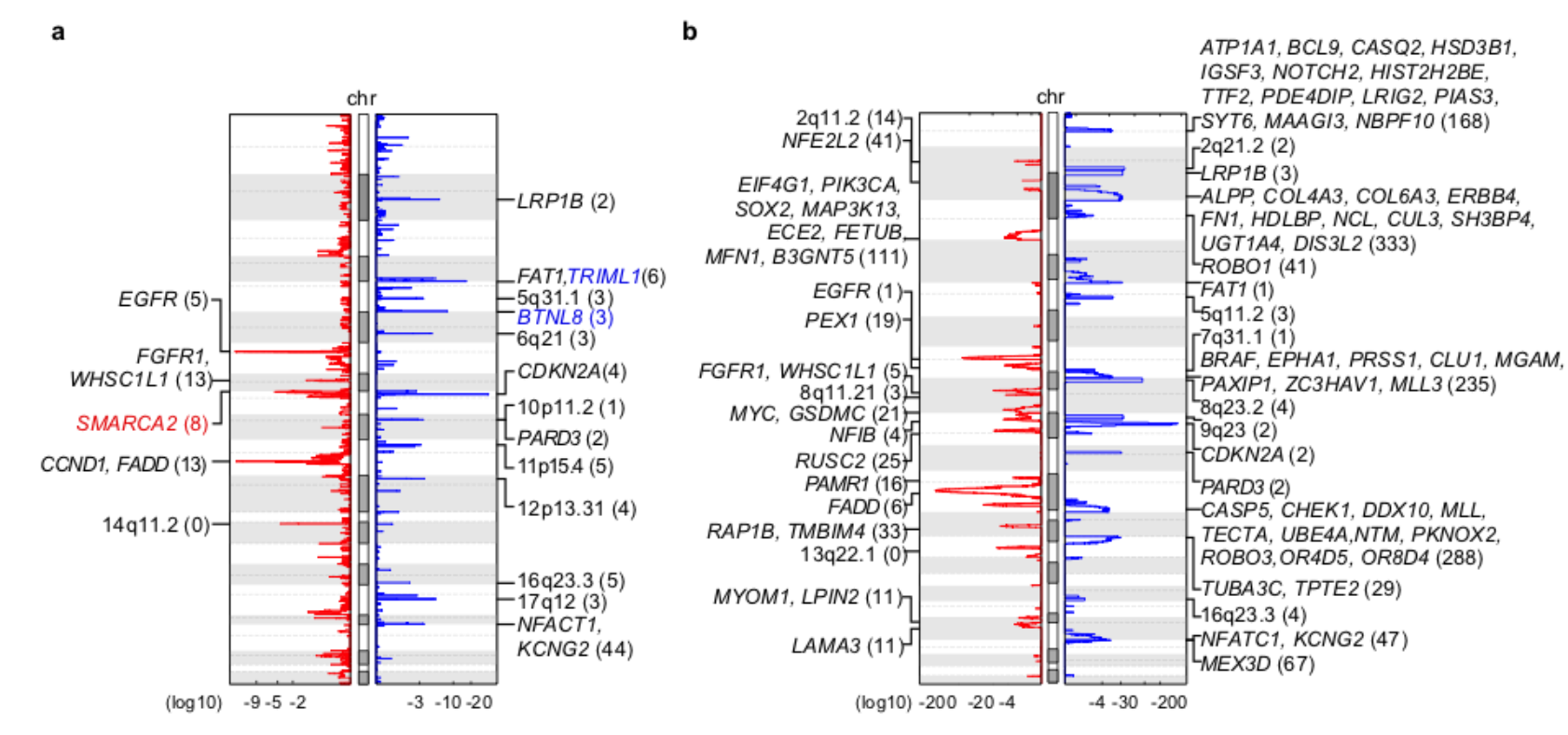
然后可以再具体看有差异的地方(染色体片段),整理展现4个基因组区域:
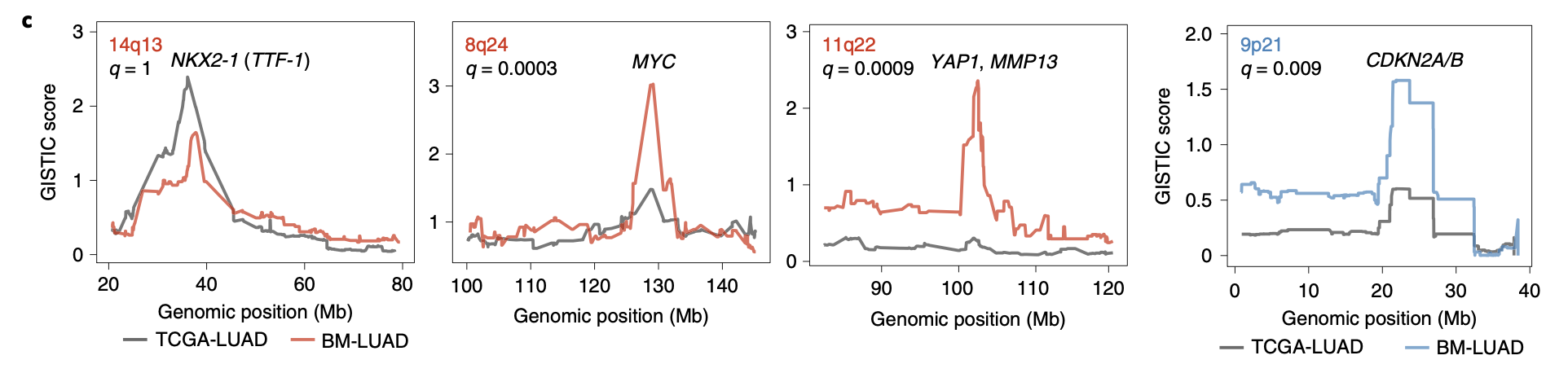
图例是: GISTIC plots of control region (NKX2-1) and candidate metastatic driver regions
可以看到 这个区域 region (NKX2-1) 在图上面是没有拷贝数差异的,但是另外的3个区域很明显在两个队列有肉眼可见的差异。有意思的是前面的差异分析散点图里面密码是4个区域,这个时候 candidate metastatic driver regions仅仅是展现了3个区域,缺了那个4q31区域,我猜想可能是这个4q31区域上面并没有很出名的基因,所以被研究者首先排除了。
最后箱线图去验证拷贝数差异区域上面的热点基因的拷贝数情况:
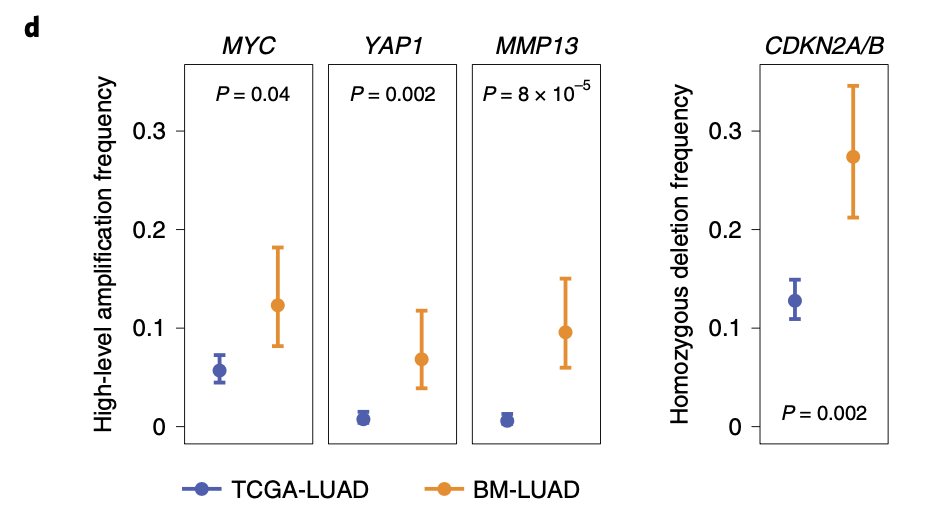
图例是:d, Frequencies of amplifications or deletions of candidate metastatic drivers, adjusted by matching weights to control for confounding.
为了让在数据分析更可信,研究者们还加入了实验验证:
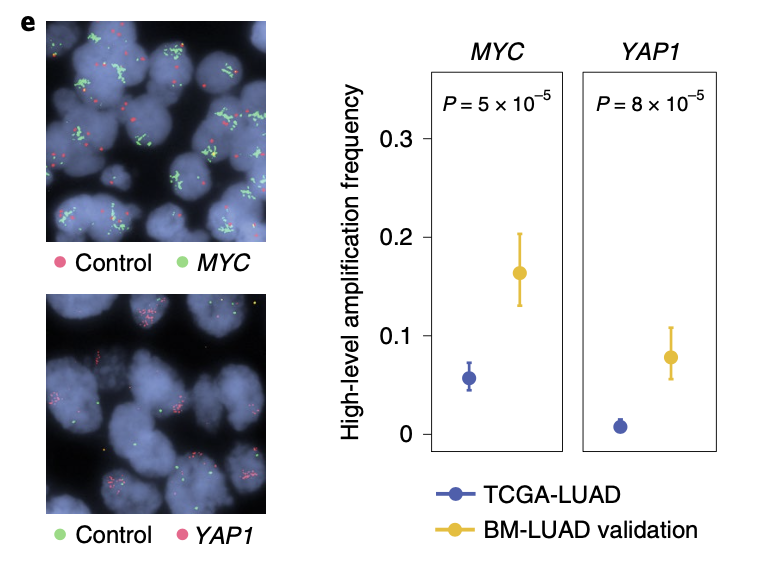
图例是:e, Frequencies of amplifications of MYC and YAP1 in the BM-LUAD validation cohort (n = 105) as determined by FISH. TCGA-LUAD was reused as the control cohort.
最后作者的结论是:Amplified candidate drivers (MYC, MMP13, YAP1) tended to occur after the divergence of the metastatic and primary tumor lineages, in agreement with positive selection of these amplifica- tions contributing to a pro-metastatic phenotype
那么问题来了,这样的数据分析如何做呢?
这个还是有点难度的, 首先你需要拿到两个队列各自的cnv信息,如果是肿瘤外显子数据,首先需要对fastq数据进行质量控制,比对到参考基因组拿到bam文件,接着使用cnvkit处理这些bam文件进行cnv分析,CNVkit的标准流程,代码如下:
# From baits and tumor/normal BAMs
ref=/home/biosoft/GATK/resources/bundle/hg38/Homo_sapiens_assembly38.fasta
bed=/home/annotation/CCDS/human/hg38.baits.bed
source activate wes
## set --method amplicon if the data from PCR
cnvkit.py batch tumor_bam/*.bam \
--normal normal_bam/*.bam \
--targets $bed \
--fasta $ref \
--output-reference my_reference.cnn --output-dir T_results
运行起来还是非常快的,这个cnv分析对每个肿瘤外显子队列都是需要测肿瘤样品也同步测序其正常对照哦,这样分析才能是可靠的。
多样本的cns文件可以合并到IGV查看,很简单的一个awk脚本即可,如下:
awk '{print FILENAME"\t"$0}' *_recal.cns |grep -v chromosome |sed 's/_recal.cns//g' |awk '{print $1"\t"$2"\t"$3"\t"$4"\t"$8"\t"$6}' >final.seg
不同队列的 final.seg 文件,都需要去走前面提到的 GISTIC流程 ,一般来说每个 cnv文件(final.seg )走了GISTIC流程 ,都会得到如下所示:
但是两个队列两个这样的图片,很难去肉眼比较,所以需要参考这个文章:《Genomic characterization of human brain metastases identifies drivers of metastatic lung adenocarcinoma》
如果你也恰好有两个队列的肿瘤外显子数据,或者有想挖掘的公共数据库里面的队列可以区分成为两个分组,我们提供这样的数据分析服务,绘制同样的4个图表:
- 肿瘤拷贝数扩增和缺失全景图
- 两个队列的cnv信息的的 GISTIC流程结果,并且差异分析散点图
- 局部差异区域可视化
- 局部差异区域基因可视化
费用1600人民币!
如果是fastq数据,需要比对到参考基因组并且找变异并且注释,我们仅仅是收取一个计算机资源的费用,800-8000元人民币(根据样品数量不同收费不一样)即可,并且提供全套代码。不管是公共数据集还是你自己的实验测序数据,一样的费用!我们会代替你这个流程,见:WES,WGS等DNA测序数据找变异流程服务