我们以PNAS杂志的一个关于AD的单细胞的数据集, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE157827 为例子,它有 21个10x样品哦,总计是 169,496 nuclei 单细胞,大家可以去下载它的文件:会得到如下所示:
32K 9 3 2020 GSM4775561_AD1_barcodes.tsv.gz
298K 9 3 2020 GSM4775561_AD1_features.tsv.gz
53M 9 3 2020 GSM4775561_AD1_matrix.mtx.gz
74K 9 3 2020 GSM4775562_AD2_barcodes.tsv.gz
298K 9 3 2020 GSM4775562_AD2_features.tsv.gz
115M 9 3 2020 GSM4775562_AD2_matrix.mtx.gz
32K 9 3 2020 GSM4775563_AD4_barcodes.tsv.gz
298K 9 3 2020 GSM4775563_AD4_features.tsv.gz
33M 9 3 2020 GSM4775563_AD4_matrix.mtx.gz
61K 9 3 2020 GSM4775564_AD5_barcodes.tsv.gz
298K 9 3 2020 GSM4775564_AD5_features.tsv.gz
70M 9 3 2020 GSM4775564_AD5_matrix.mtx.gz
16K 9 3 2020 GSM4775565_AD6_barcodes.tsv.gz
298K 9 3 2020 GSM4775565_AD6_features.tsv.gz
5.6M 9 3 2020 GSM4775565_AD6_matrix.mtx.gz
下游处理的时候这些文件时候,一定要保证每个样品的 3个文件同时存在,而且在同一个文件夹下面。 示例代码是:
rm(list=ls())
options(stringsAsFactors = F)
library(Seurat)
sce1 <- CreateSeuratObject(Read10X('../10x-results/WT/'),
"wt")
重点就是 Read10X 函数读取 文件夹路径,比如:../10x-results/WT/ ,保证文件夹下面有3个文件。
其实如果样本量并不大,可以自己手动修改,如下所示,每个文件夹里面都是3个文件,而且文件名必须很纯净,保证是一样的:
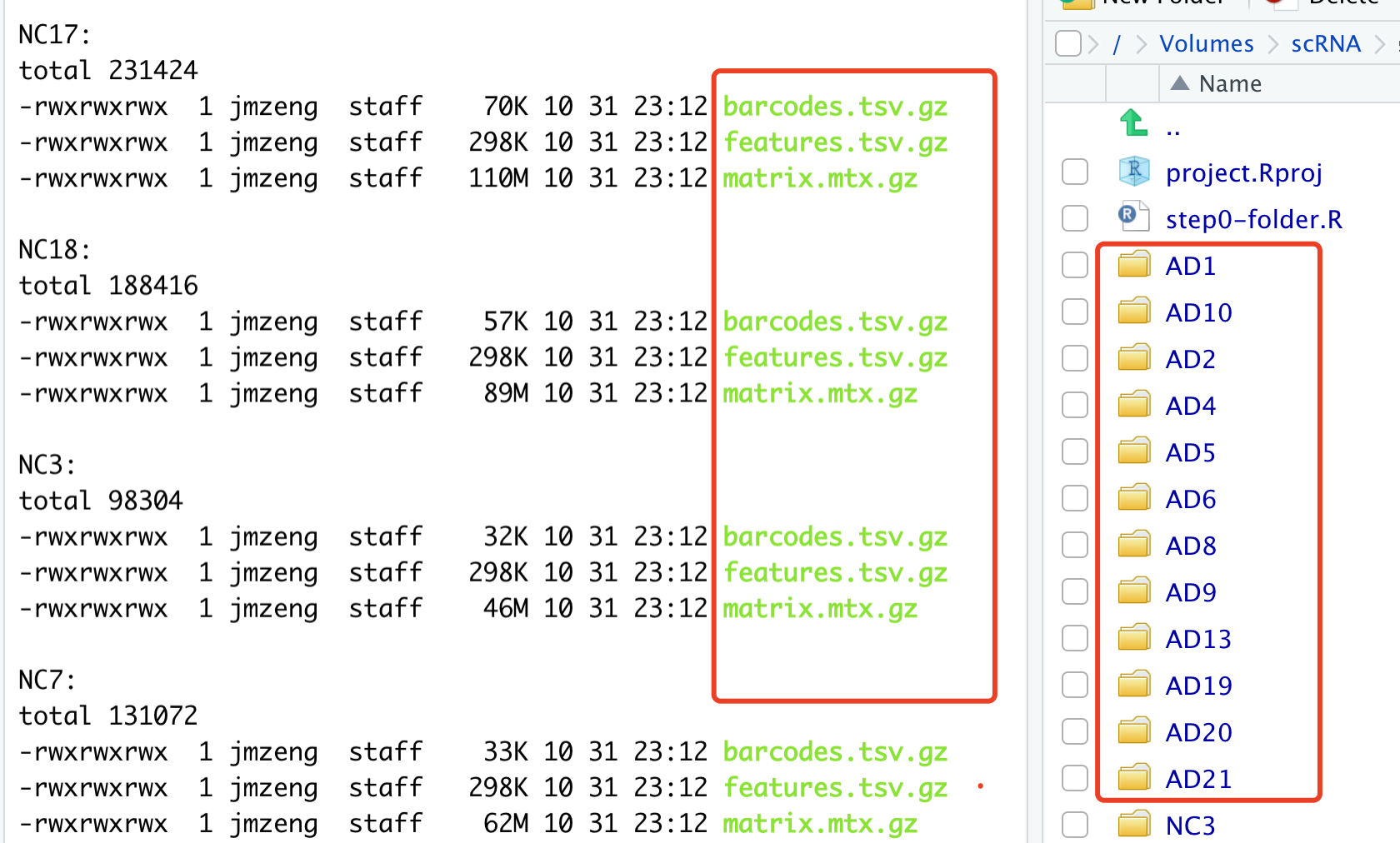
但是如果样品数量很多,其实就可以使用代码批量进行修改啦!
fs=list.files('./','features.tsv.gz')
fs
samples1=gsub('features.tsv.gz','',fs)
samples1
library(stringr)
samples2=str_split(samples1,'_',simplify = T)[,2]
samples2
lapply(1:length(samples2), function(i){
x=samples2[i]
y=samples1[i]
dir.create(x,recursive = T)
file.copy(from=paste0(y,'features.tsv.gz'),
to=file.path(x, 'features.tsv.gz' ))
file.copy(from=paste0(y,'matrix.mtx.gz'),
to= file.path(x, 'matrix.mtx.gz' ) )
file.copy(from=paste0(y,'barcodes.tsv.gz'),
to= file.path(x, 'barcodes.tsv.gz' ))
})
赶快去试试看吧!
接下来就可以对这个文件夹进行批量读入啦,示例代码是:
dir='../GSE157827_RAW/outputs'
samples=list.files( dir )
samples
sceList = lapply(samples,function(pro){
#pro=samples[1]
folder=file.path(dir ,pro )
print(pro)
print(folder)
print(list.files(folder))
sce=CreateSeuratObject(counts = Read10X(folder),
project = pro )
return(sce)
})
是不是很方便啊!
其实好多年以前我在教程:使用seurat3的merge功能整合8个10X单细胞转录组样本 和 seurat3的merge功能和cellranger的aggr整合多个10X单细胞转录组对比 就给出了后续R代码读取10x单细胞转录组数据的3个文件的表达矩阵。
大家在下面的文章里面可以搜索到10x单细胞转录组数据的文章公布在geo数据库的链接:
如果你对单细胞数据分析还没有基础认知,可以看基础10讲: