这个策略目前应用蛮广泛的, 比如文章《A Novel Inflammatory lncRNAs Prognostic Signature for Predicting the Prognosis of Low-Grade Glioma Patients》就是提取TCGA和CGGA共有的炎症基因mRNA和lncRNA的表达数据后,采用Pearson法分析lncRNA与LGGs炎症相关基因的相关性,相关系数为| r| > 0.7的lncRNA视为炎症相关lncRNA。当然了,也可以提取免疫相关mRNA基因,自噬的,细胞焦亡的,缺氧的。
总之,一个表达量矩阵里面起码有两万个mRNA基因,接近两万个lncRNA基因,如果他们进行相关性计算,会产生4亿个相互关系,哪怕是使用 相关系数为| r| > 0.9 也仍然是有数量太多的 mRNA和lncRNA 配对关系。所以大家会倾向于使用生物学功能基因集,比如提取免疫相关mRNA基因,自噬的,细胞焦亡的,缺氧的。相当于是人为把基因数量搞小。
我们现在以airway这个转录组测序表达量矩阵,来演示如何针对 特定mRNA基因的相关性lncRNA计算。
首先获取表达量矩阵
#BiocManager::install("airway")
suppressPackageStartupMessages( library( "GEOquery" ) )
suppressPackageStartupMessages( library( "airway" ) )
library("airway")
data(airway)
airway
as.data.frame(colData(airway))
summary(colSums(assay(airway))/1e6)
metadata(rowRanges(airway))
ensembl_matrix=assay(airway)
ensembl_matrix[1:4,1:4]
如下所示:
> ensembl_matrix[1:4,1:4]
SRR1039508 SRR1039509 SRR1039512 SRR1039513
ENSG00000000003 679 448 873 408
ENSG00000000005 0 0 0 0
ENSG00000000419 467 515 621 365
ENSG00000000457 260 211 263 164
> dim(ensembl_matrix)
[1] 64102 8
可以看到, 是 6万多个基因的表达量矩阵,在18个样品里面。但是,众所周知人类仅仅是2万个左右的蛋白质编码基因,所以其它都是非编码。需要区分开来。
借助AnnoProbe包里面的gtf文件信息来区分 mRNA和lncRNA
需要把区分 mRNA和lncRNA拆分成为两个表达量矩阵,方便后续进行相关性计算。
library(AnnoProbe)
gs=annoGene(rownames(ensembl_matrix),'ENSEMBL','human')
head(gs)
tail(sort(table(gs$biotypes)))
pd_genes=gs[gs$biotypes=='protein_coding',1]
non_genes=gs[gs$biotypes !='protein_coding',1]
pd_matrix=ensembl_matrix[rownames(ensembl_matrix) %in% pd_genes,]
non_matrix=ensembl_matrix[rownames(ensembl_matrix) %in% non_genes,]
save(non_matrix,pd_matrix,
file = 'input.Rdata')
如下所示:
> head(gs)
SYMBOL biotypes ENSEMBL chr start end
1 DDX11L1 transcribed_unprocessed_pseudogene ENSG00000223972 chr1 11869 14409
2 WASH7P unprocessed_pseudogene ENSG00000227232 chr1 14404 29570
4 MIR1302-2HG lncRNA ENSG00000243485 chr1 29554 31109
6 FAM138A lncRNA ENSG00000237613 chr1 34554 36081
7 OR4G4P unprocessed_pseudogene ENSG00000268020 chr1 52473 53312
8 OR4G11P transcribed_unprocessed_pseudogene ENSG00000240361 chr1 57598 64116
> as.data.frame(tail(sort(table(gs$biotypes))))
Var1 Freq
1 misc_RNA 1699
2 snRNA 1805
3 unprocessed_pseudogene 2102
4 processed_pseudogene 9739
5 lncRNA 12994
6 protein_coding 19354
也可以仅仅是考虑 protein_coding 和lncRNA ,我前面的代码是区分 protein_coding和所有的非编码。
相关性计算
首先检查拆分后的 protein_coding和所有的非编码两个独立的表达量矩阵各自的特性
rm(list = ls())
options(stringsAsFactors = F)
load( file = 'input.Rdata')
cg=tail(names(sort(apply(non_matrix, 1, mad))),5000)
non_matrix=non_matrix[cg,]
cg=tail(names(sort(apply(pd_matrix, 1, mad))),5000)
pd_matrix=pd_matrix[cg,]
# log 会影响表达量相关性
cor(as.numeric(non_matrix[1,]),
as.numeric(pd_matrix[1,]))
cor(log(as.numeric(non_matrix[1,])),
log(as.numeric(pd_matrix[1,])))
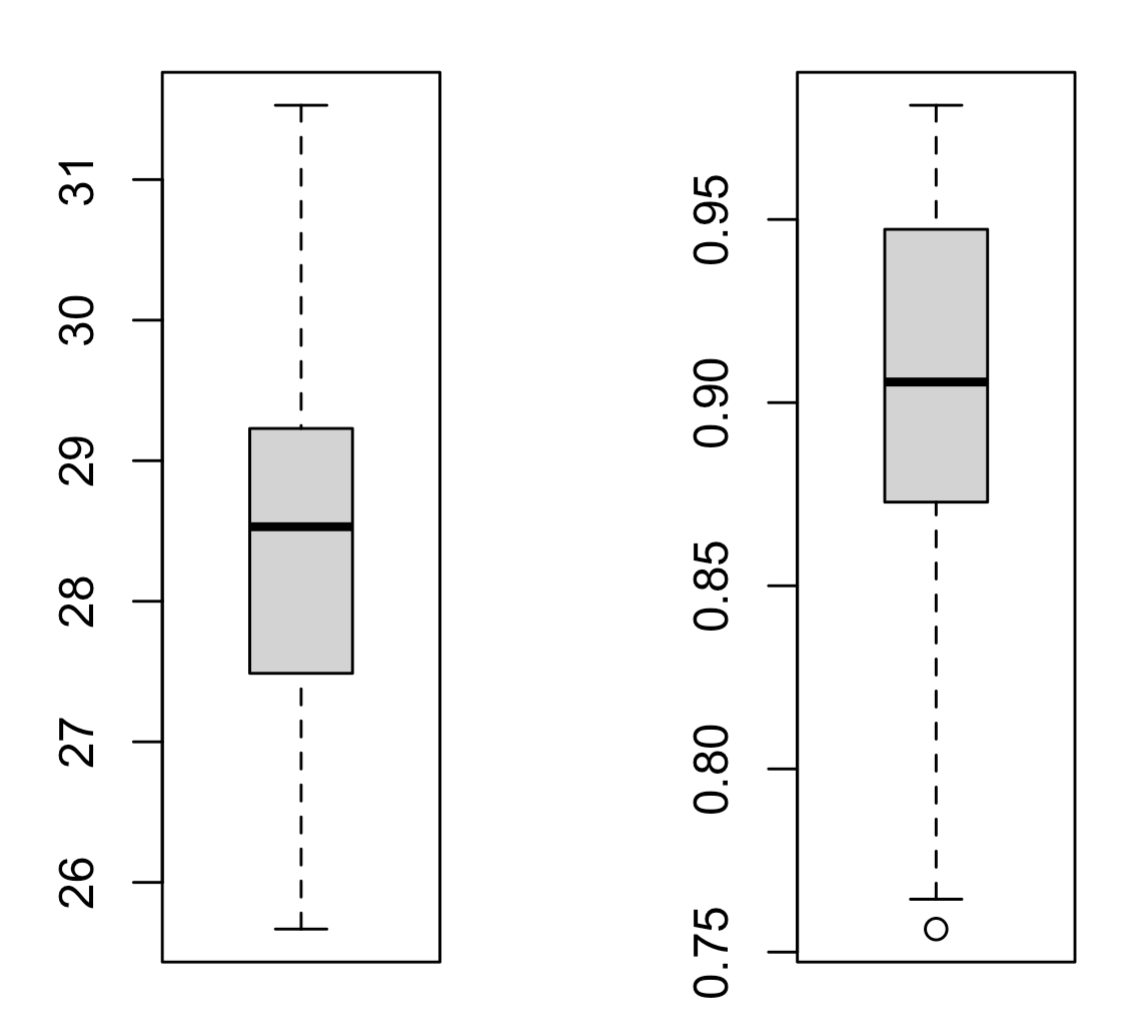
par(mfrow=c(1,2))
boxplot(colSums(pd_matrix)/1000000)
boxplot(colSums(non_matrix)/1000000)
可以看到,编码基因表达量平均是非编码的30倍以上,如下所示:
现在可以计算相关性:
library(edgeR)
non_matrix=log2(edgeR::cpm(non_matrix)+1)
pd_matrix=log2(edgeR::cpm(pd_matrix)+1)
cor(as.numeric(non_matrix[1,]),
as.numeric(pd_matrix[1,]))
pd_matrix[1:4,1:4]
non_matrix[1:4,1:4]
# 单独的个例来说,相关性提高了。
table(phe)
dat=rbind(pd_matrix,non_matrix)
# Unfortunately, the function cor() returns only the correlation coefficients between variables
cor_dat=cor(t(dat))
cor_df=cor_dat[1:nrow(pd_matrix),(nrow(pd_matrix)+1):(nrow(pd_matrix)+nrow(non_matrix))]
round(cor_df[1:4,1:4], 2)
简单看看相关性的结果:
library(reshape2)
df=melt(cor_df)
head(df)
dim(df)
table(abs(df$value) > 0.9)
table(abs(df$value) > 0.99)
df_filter=df[abs(df$value) > 0.99,]
head(df_filter)
tail(df_filter)
dim(df)
head(df)
sort(table(as.character(df_filter[,2])))
sort(table(as.character(df_filter[,1])))
如下所示:
> table(abs(df$value) > 0.9)
FALSE TRUE
24845266 154734
> table(abs(df$value) > 0.99)
FALSE TRUE
24999890 110
> head(df_filter)
Var1 Var2 value
15938727 CPT1A AC079949.5 -0.9902789
18855267 MMP1 AC097634.1 -0.9932103
18855403 MYEOV AC097634.1 -0.9904240
18855421 SYT9 AC097634.1 -0.9933735
18855611 BDNF AC097634.1 0.9915814
18855973 STAMBPL1 AC097634.1 -0.9915057
> tail(df_filter)
Var1 Var2 value
23928887 MAGEA6 LINC00461 0.9906351
23928983 SBSPON LINC00461 0.9903057
24521441 CDA DUBR 0.9911254
24523994 CREG1 DUBR 0.9924729
24524456 SLC39A8 DUBR 0.9920506
24849508 SLC9A3R2 OIP5-AS1 -0.9915939
因为这个表达量矩阵仅仅是18个样品,而且是不同的生物学分组, 所以这里面的相关性是有问题的。
也可以具体去可视化某个相关性:
# MAGEA6 LINC00461 0.9906351
library(ggstatsplot)
colnames(dat)
dat=as.data.frame(t(dat))
colnames(dat)
ggscatterstats(data = dat ,
y = LINC00461,
x = MAGEA6,
centrality.para = "mean",
margins = "both",
xfill = "#CC79A7",
yfill = "#009E73",
marginal.type = "histogram",
title = "Relationship between a and b")
其实很有意思:
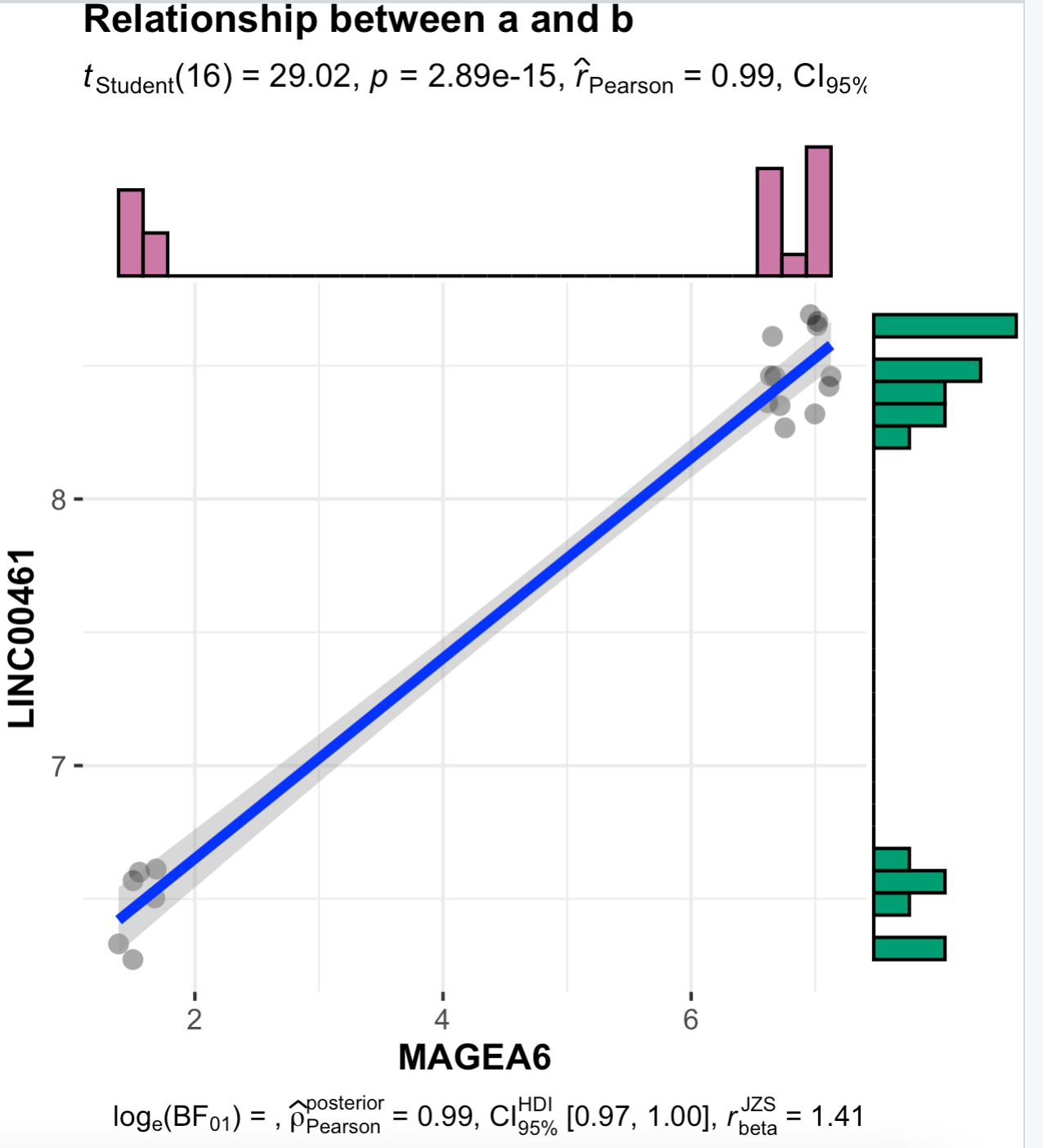
这个时候的相关性其实是有问题的哦,
如果你同时需要p值
另外一个方案:
# http://www.sthda.com/english/wiki/correlation-matrix-a-quick-start-guide-to-analyze-format-and-visualize-a-correlation-matrix-using-r-software
library("Hmisc")
my_data=t(dat)
dim(my_data)
# Error: vector memory exhausted (limit reached?)
res2 <- rcorr(my_data)
res2
# Extract the correlation coefficients
res2$r
# Extract p-values
res2$P
library(psych)
my_data=t(dat)
dim(my_data)
# Error: vector memory exhausted (limit reached?)
res2 <- corr.test(my_data)
还有更高级可视化
不过,我觉得可视化都是纸老虎,好的结果才是重心!