曾经(大约是2010附近)普通的bulk的转录组测序跟如今的单细胞转录组一样火爆,是个样品就去测序,都不会理会类似的实验设计是否有已经发表的文章。都妄想用经费堆,去摘低垂的果实,所以大量数据烂在手上,拖到后面就越来越难以发表,能捡到个普通杂志发表出去就谢天谢地了。
比如发表在《genes & genomics》这个期刊(影响因子1分左右)的 2016的文章:《Transcriptome analysis of non-small cell lung cancer and genetically matched adjacent normal tissues identifies novel prognostic marker genes》,链接是 https://link.springer.com/article/10.1007/s13258-016-0492-5 ,就是一个超级普通的bulk的转录组测序数据。
他们自己的课题设计其实仅仅是做了 10 pairs of genetically matched transcriptome (NSCLC and adjacent normal tissues obtained from 10 patients) ,其实这样的数据本来就可以直接去TCGA数据库提取了,完全没有必要自己招募病人自己花钱测序。数量级上不去,分析也是平平无奇,所以作者得结合两个公共数据:71 pairs (GSE40419) and 58 pairs (TCGA-LUAD),
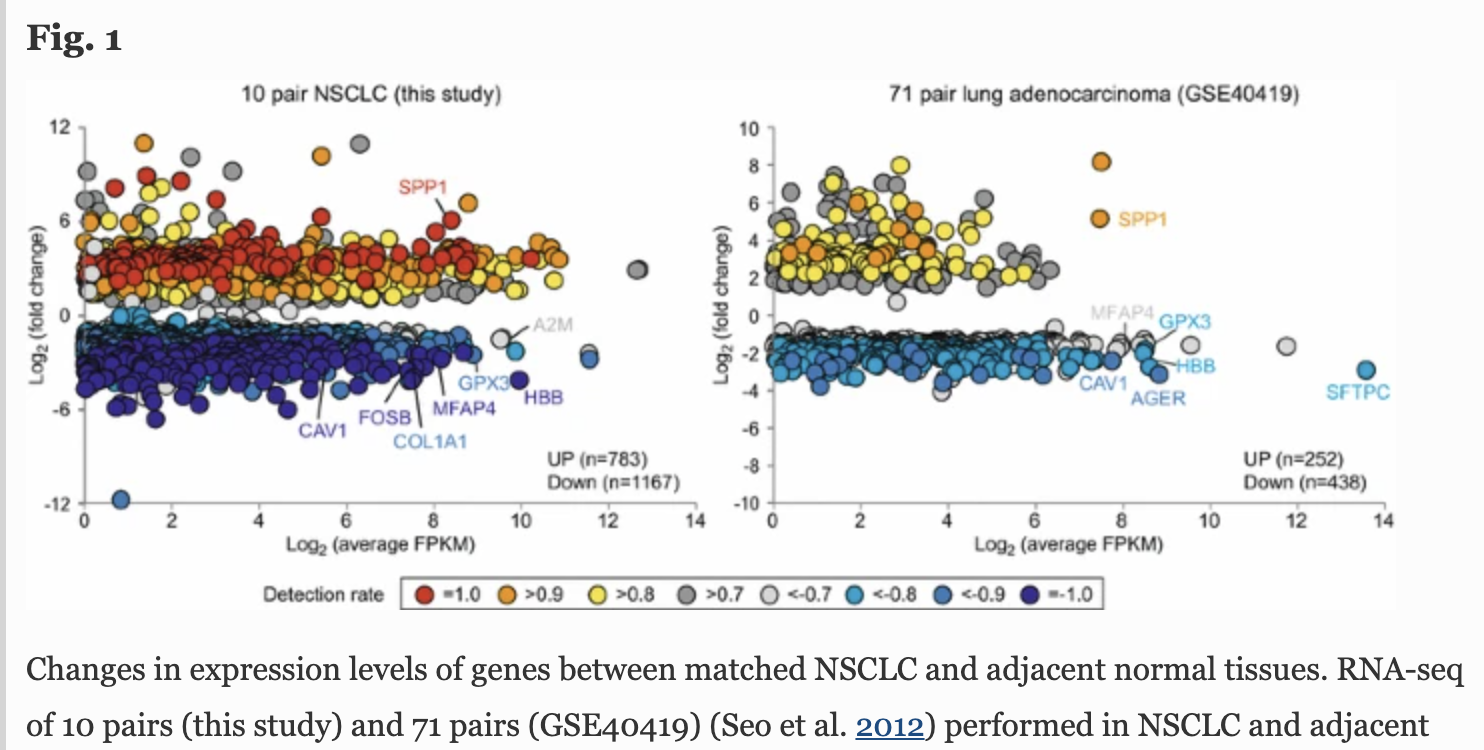
这样就有3次差异分析,可以取交集看韦恩图:
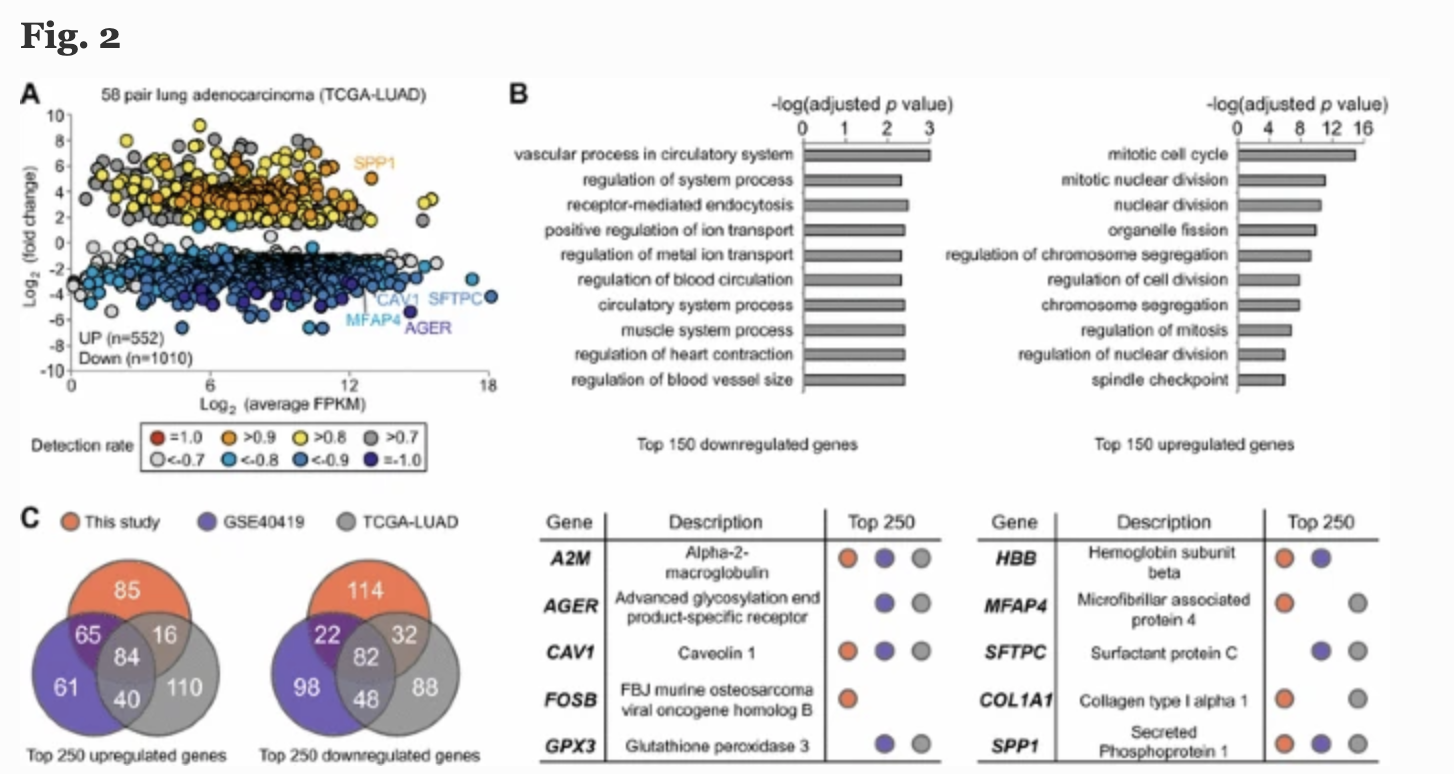
普通的转录组数据分析超级简单
有意思的是都2021了仍然有人转录组走tophat加cufflinks流程,过时了,我在2015年逛biostar论坛的时候,看到了这样的表述:
Tophat 首次被发表已经是6年前
Cufflinks也是五年前的事情了
Star的比对速度是tophat的50倍,hisat更是star的1.2倍。
stringTie的组装速度是cufflinks的25倍,但是内存消耗却不到其一半。
Ballgown在差异分析方面比cuffdiff更高的特异性及准确性,且时间消耗不到cuffdiff的千分之一
Bowtie2+eXpress做质量控制优于tophat2+cufflinks和bowtie2+RSEM
Sailfish更是跳过了比对的步骤,直接进行kmer计数来做QC,特异性及准确性都还行,但是速度提高了25倍
kallisto同样不需要比对,速度比sailfish还要提高5倍!!!
当时各路大神就建议大家抛弃传统的tophat加cufflinks流程,毕竟其作者都说它过时了,起码可以替换成为:hisat2+stringtie+ballgown流程啊!
如果你看到有人还在使用tophat加cufflinks流程来处理转录组数据,也不要急于嘲讽,有可能是他们的数据本来就是五六年前的,或者给他们服务的公司仍然是使用过时的流程而已。
但是我们的B站免费NGS数据处理视频课程就不会如此过时,因为常规ngs组学早在2015就定型了,我们的视频课程大多在2018年前后制作,已经组建了微信交流群的有下面这些:
- 免费视频课程《RNA-seq数据分析》
- 免费视频课程《WES数据分析》
- 免费视频课程《ChIP-seq数据分析》
- 免费视频课程《ATAC-seq数据分析》
- 免费视频课程《TCGA数据库分析实战》
- 免费视频课程《甲基化芯片数据分析》
- 免费视频课程《影像组学教学》
- 免费视频课程《LncRNA-seq数据》
- 免费视频课程《GEO数据挖掘》
- 肿瘤基因测序
最后,既然是肿瘤病人的测序数据,分析到最后,肯定是画龙点睛一下,添加生存分析,使用 lung cancer microarray datasets (GSE41271, GSE37745, and GSE4573) ,就可以说明自己的测序数据分析得到的基因是有临床意义的。
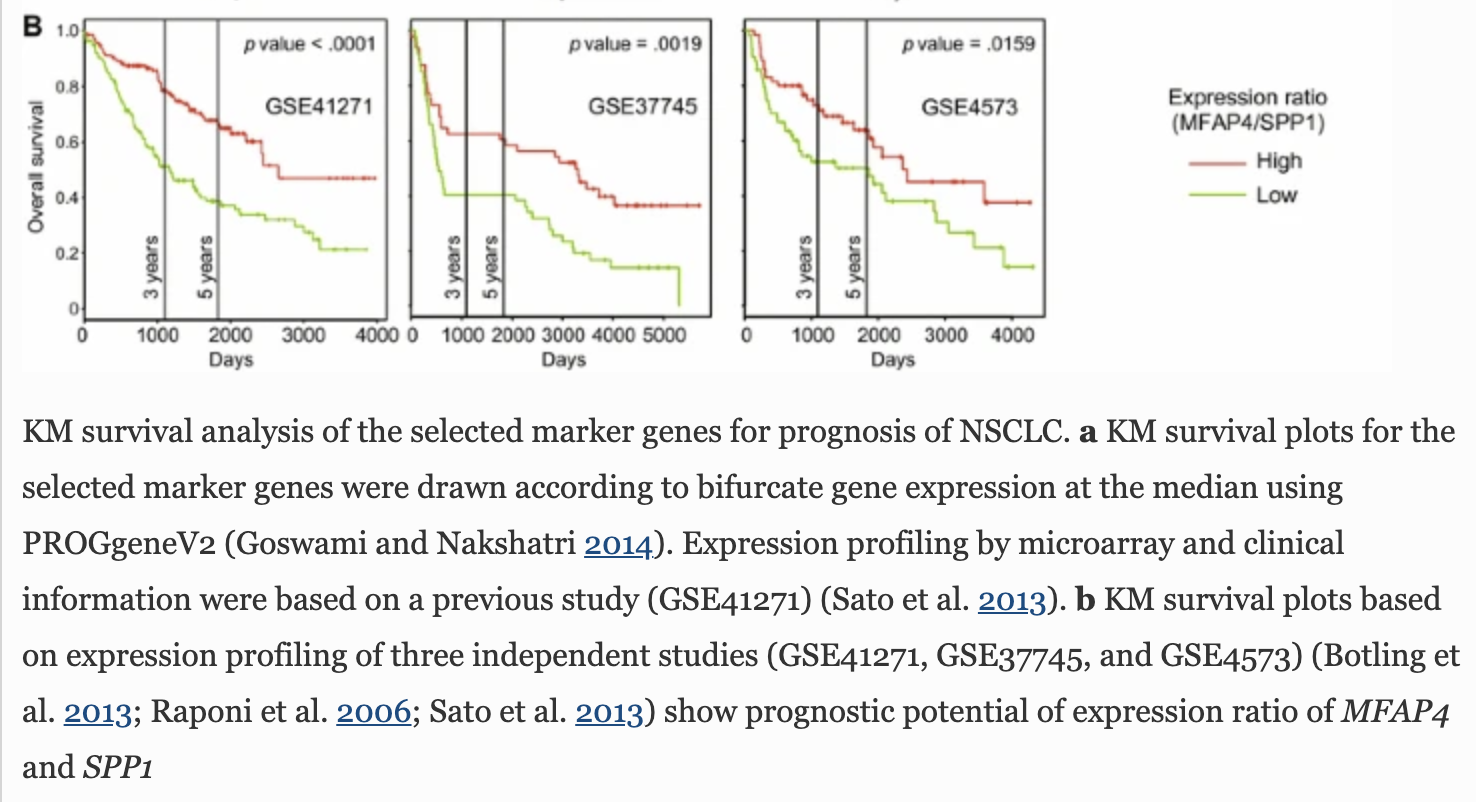
我在生信技能树多次分享过生存分析的细节;
- 人人都可以学会生存分析(学徒数据挖掘)
- 学徒数据挖掘之谁说生存分析一定要按照表达量中位值或者平均值分组呢?
- 基因表达量高低分组的cox和连续变量cox回归计算的HR值差异太大?
- 学徒作业-两个基因突变联合看生存效应
- TCGA数据库里面你的基因生存分析不显著那就TMA吧
- 对“不同数据来源的生存分析比较”的补充说明
- 批量cox生存分析结果也可以火山图可视化
- 既然可以看感兴趣基因的生存情况,当然就可以批量做完全部基因的生存分析
- 多测试几个数据集生存效应应该是可以找到统计学显著的!
- 我不相信kmplot这个网页工具的结果(生存分析免费做)
- 为什么不用TCGA数据库来看感兴趣基因的生存情况
- 200块的代码我的学徒免费送给你,GSVA和生存分析
- 集思广益-生存分析可以随心所欲根据表达量分组吗
- 生存分析时间点问题
- 寻找生存分析的最佳基因表达分组阈值
- apply家族函数和for循环还是有区别的(批量生存分析出图bug)
- TCGA数据库生存分析的网页工具哪家强
- KM生存曲线经logRNA检验后也可以计算HR值
生存分析是目前肿瘤等疾病研究领域的点睛之笔!