最近遇到了很烦人的事情,就是一口气做了三百多个单细胞转录组项目(主要是GEO上面的),然后代码一直在修正和完善,尤其是可视化。并不是说自己要创造什么炫酷的可视化方式,以前我们做了一个投票:[可视化单细胞亚群的标记基因的5个方法](https://mp.weixin.qq.com/s/enGx9_Sv5wKLdtygL7b4Jw),下面的5个基础函数相信大家都是已经烂熟于心了:
- VlnPlot(pbmc, features = c(“MS4A1”, “CD79A”))
- FeaturePlot(pbmc, features = c(“MS4A1”, “CD79A”))
- RidgePlot(pbmc, features = c(“MS4A1”, “CD79A”), ncol = 1)
- DotPlot(pbmc, features = unique(features)) + RotatedAxis()
- DoHeatmap(subset(pbmc, downsample = 100), features = features, size = 3)
就是这最基础的函数,用好就挺不容易的。简单的降维聚类分群,可以参考前面的例子:人人都能学会的单细胞聚类分群注释 ,我们演示了第一层次的分群。然后就需要挑选合适的基因在不同亚群里面展现一下,代码如下所示:
library(stringr)
genes_to_check=str_to_title(unique(genes_to_check))
genes_to_check
p <- DotPlot(sce.all, features = genes_to_check,
assay='RNA' ,group.by = 'celltype' ) + coord_flip()
p
这个最基础的DotPlot出图会比较难看,因为各个细胞亚群的名字拥挤到了一起!
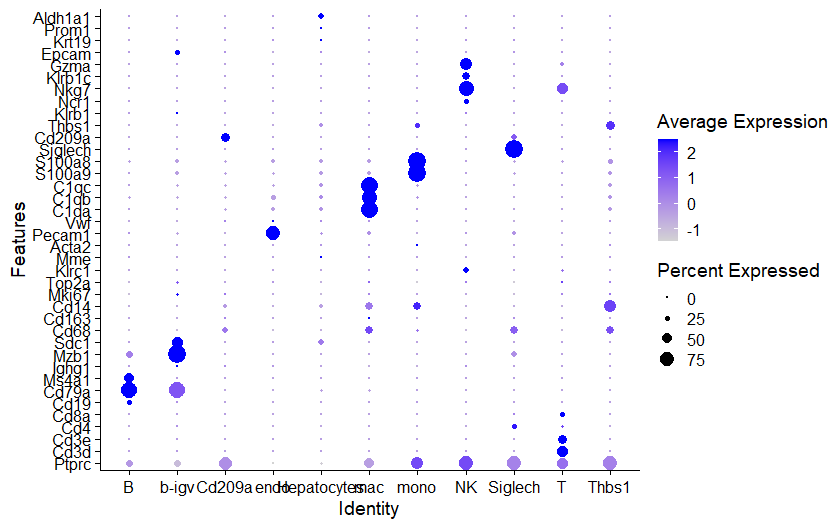
学过ggplot语法的小伙伴都知道如何调整,设置X坐标轴的字符串的方向即可:
th=theme(axis.text.x = element_text(angle = 45,
vjust = 0.5, hjust=0.5))
library(stringr)
genes_to_check=str_to_title(unique(genes_to_check))
genes_to_check
p <- DotPlot(sce.all, features = genes_to_check,
assay='RNA' ,group.by = 'celltype' ) + coord_flip() #+th
p
如下所示,可以很清楚的看清楚各个细胞亚群啦!
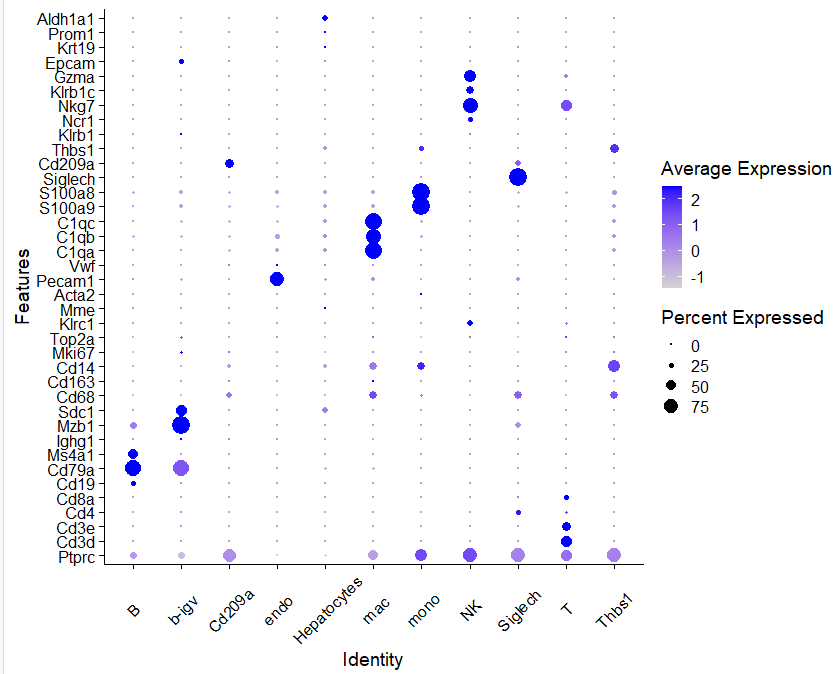
问题就是,每次我都懒得写这个代码,临时去谷歌搜索,不小心搜到了这样的一个代码:
p_all_markers=DotPlot(sce.all, features = genes_to_check,
assay='RNA' ,group.by = 'singleR' ) +
coord_flip()+ scale_y_discrete(guide = guide_axis(n.dodge = 2)) +
NULL
看起来也挺有意思的,效果如下所示:
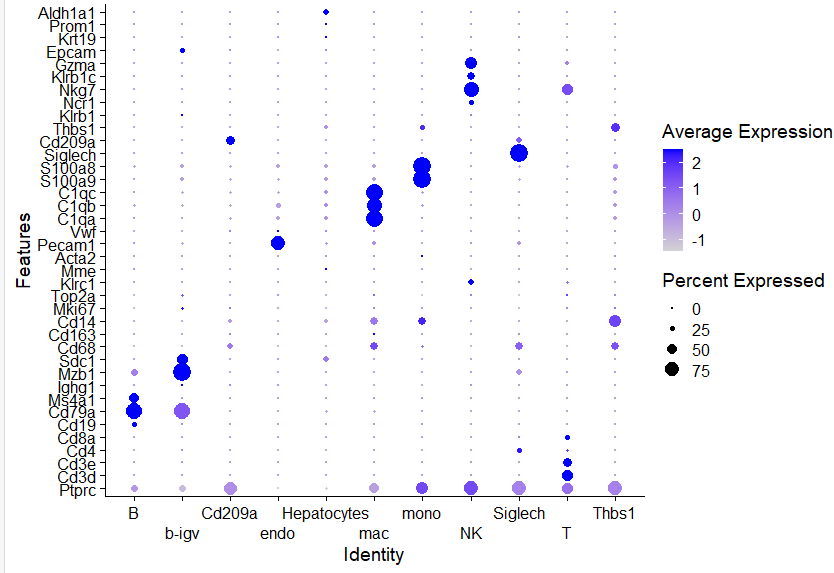
如果你感兴趣单细胞转录组数据处理,可以去follow我们的两个b站单细胞栏目,持续更新半年,基本上涵盖了大家需要的技能:
- https://www.bilibili.com/video/BV1DK4y1X7bb/ 更新至第8篇,「生信技能树」100个单细胞文献解读(8/100)
- https://www.bilibili.com/video/BV19Q4y1R7cu/ section 3已更新,「生信技能树」单细胞公开课2021
如果是简单的降维聚类分群,可以参考前面的例子:人人都能学会的单细胞聚类分群注释 ,我们演示了第一层次的分群。
如果你对单细胞数据分析还没有基础认知,可以看基础10讲: