前面我们演示了:泛癌分析时候的大样本量多分组建议选择tSNE而不是PCA,整合全部的33种癌症的仅仅是蛋白质编码基因的表达量矩阵,进行降维聚类分群可以看到并不是严格的各个癌症泾渭分明。
- 其中很明显乳腺癌区分成为了不同亚群,应该是对应着不同的分子分型,尤其是三阴性或者说basal-like的,跟乳腺癌主群区分很开。
- 而KICH,KIRC,KIRP 这3种肾癌,互相之间纠缠不清。
- LUAD和LUSC两个肺癌也是有部分混淆。
- 其中COAD,READ,STAD主要的消化道癌症被聚类在了一起,也许是因为他们的性质过于类似吧。
- 让我意外的是THCA居然也被拆分成为了两个主要的小群,这个是我的知识盲区了。(发朋友圈后有小伙伴告诉我因为这个癌症也是区分为迥异的鳞癌和腺癌)
前面的流程是自己把一万多个病人样品矩阵,进行了合并后,从一万六基因里面挑选top1000的方差基因后,自己走PCA函数和tSNE函数,也进行了kmeans和dbscan的聚类分群。总体来说,结果还可以。
但是这个降维聚类分群步骤,是不是让大家想起来了我们最近一直强调的单细胞转录组数据分析流程。仍然是可以参考前面的例子:人人都能学会的单细胞聚类分群注释 ,很简单的几个函数连起来使用即可!让我们来演示一下吧!
首先 合并全部癌症表达量矩阵
仍然是需要查看前面的流程::居然有如此多种癌症(是时候开启pan-cancer数据挖掘模式),我们把全部的TCGA的33种癌症的表达量矩阵区拆分成为蛋白编码基因和非编码基因这两个不同的表达量矩阵,并且保存成为了rdata文件。所以接下来,载入全部的rdata文件, 挑选全部的表达量矩阵共有基因即可:
fs=list.files('Rdata/',pattern = 'htseq_counts')
fs
# 首先加载每个癌症的蛋白编码基因表达量矩阵
# 成为一个 list
dat_list = lapply(fs, function(x){
# x=fs[1]
pro=gsub('.htseq_counts..Rdata','',x)
print(pro)
load(file = file.path('Rdata/',x))
# 因为要走单细胞 seurat流程,所以无需处理表达量矩阵
# 直接最原始的counts即可
# dat=log2(edgeR::cpm(pd_mat)+1)
dat=pd_mat
return(dat)
})
# 最关键的代码是:
tmp=table(unlist(lapply(dat_list, rownames)))
cg=names(tmp)[tmp==33]
length(cg)
# 在33个癌症都存在的蛋白编码基因是一万六千个
all_tcga_matrix=do.call(cbind,
lapply(dat_list, function(x){
x[cg,]
}))
dim(all_tcga_matrix)
group=rep(gsub('TCGA-','',gsub('.htseq_counts..Rdata','',fs)),
unlist(lapply(dat_list, ncol)))
table(group)
千万要看清楚我们是如何合并33个表达量矩阵的哦,每个矩阵都是1.6万个基因数量不等,所以首先需要对全部的表达量矩阵的全部基因名字取交集!
可以看到我们的表达量矩阵其实是传统转录组的:
> all_tcga_matrix[1:4,1:2]
TCGA-OR-A5JP-01A TCGA-OR-A5JG-01A
A1BG 25 5
A1CF 0 3
A2M 17253 16200
A2ML1 24 31
如果是单细胞表达量矩阵,里面应该是97%都是0,但是我们这个tcga的表达矩阵,0就非常少了。
走Seurat包自带的降维聚类分群
虽然是tcga的表达量矩阵,并不是真正的单细胞,不过,Seurat包自带的降维聚类分群并不在乎这个,只要是合格的表达量矩阵就没有问题,代码如下:
library(Seurat)
sce=CreateSeuratObject(counts = all_tcga_matrix)
sce
group=as.data.frame(group)
rownames(group)=colnames(sce)
sce=AddMetaData(sce,group)
table(sce$group)
# 这里并不是真正的单细胞表达量矩阵,所以无需过滤
sce.all.filt=sce
dim(sce.all.filt)
sce.all.filt <- NormalizeData(sce.all.filt, normalization.method = "LogNormalize",
scale.factor = 10000)
GetAssay(sce.all.filt,assay = "RNA")
sce.all.filt = FindVariableFeatures(sce.all.filt)
sce.all.filt = ScaleData(sce.all.filt, vars.to.regress = c("nFeature_RNA" ))
sce.all.filt = RunPCA(sce.all.filt, npcs = 20)
sce.all.filt = RunTSNE(sce.all.filt, npcs = 20)
sce.all.filt = RunUMAP(sce.all.filt, dims = 1:10)
sce=sce.all.filt
sce <- FindNeighbors(sce, dims = 1:15)
sce <- FindClusters(sce, resolution = 0.2)
table(sce@meta.data$RNA_snn_res.0.2)
进行可视化:
library(patchwork)
p1=DimPlot(sce,reduction = "tsne",label=T,repel = T,
group.by ='group')
p2=DimPlot(sce,reduction = "umap",label=T,repel = T,
group.by ='group')
p1+p2
library(patchwork)
p1=DimPlot(sce,reduction = "tsne",label=T,repel = T )
p2=DimPlot(sce,reduction = "umap",label=T,repel = T )
p1+p2
这个时候的可视化可以很清楚看到了 tsne 和 umap的差异 :
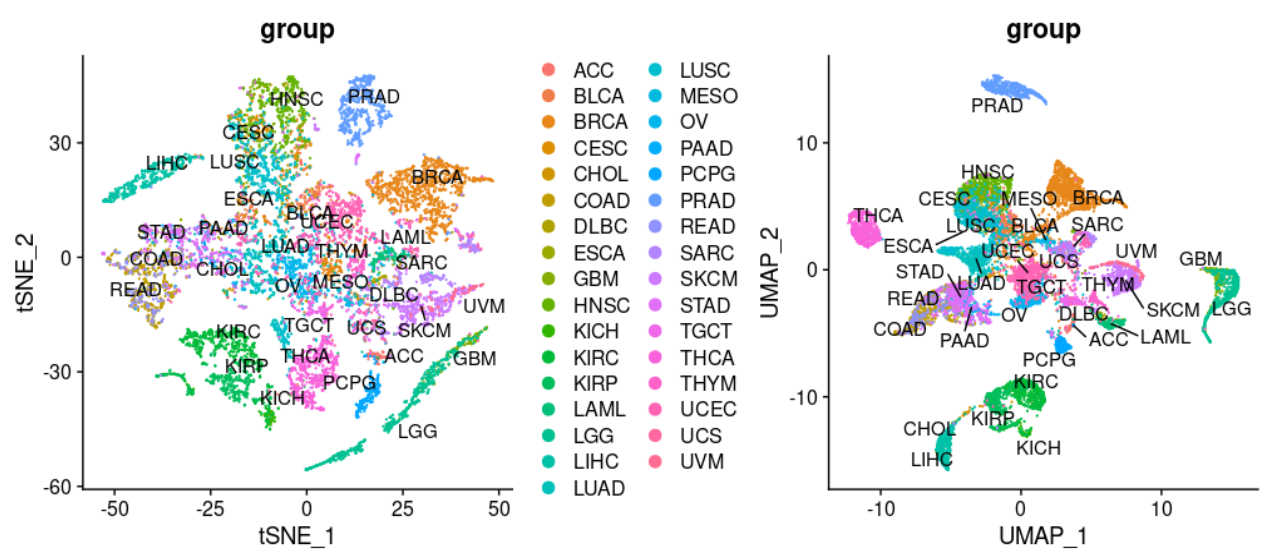
umap会把同一个亚群压缩的更密实,不同亚群会让它尽可能距离远一点,所以可以看到umap里面不同癌症类型的名字也会重叠,需要使用,repel = T这样的辅助线哦。而且可以看到我们的表达量矩阵里面可以比较明显的区分不同癌症,也算是达到了前面的流程是自己把一万多个病人样品矩阵,进行了合并后,从一万六基因里面挑选top1000的方差基因后,自己走PCA函数和tSNE函数,也进行了kmeans和dbscan的聚类分群的结果。
也就是说,所谓的单细胞seurat流程,其实是可以自己写代码完成的!
当然了,seurat有非常多方便的函数工具,初学者如果能使用当然了尽可能推荐大家使用。比如调整分群的分辨率,得到如下所示:
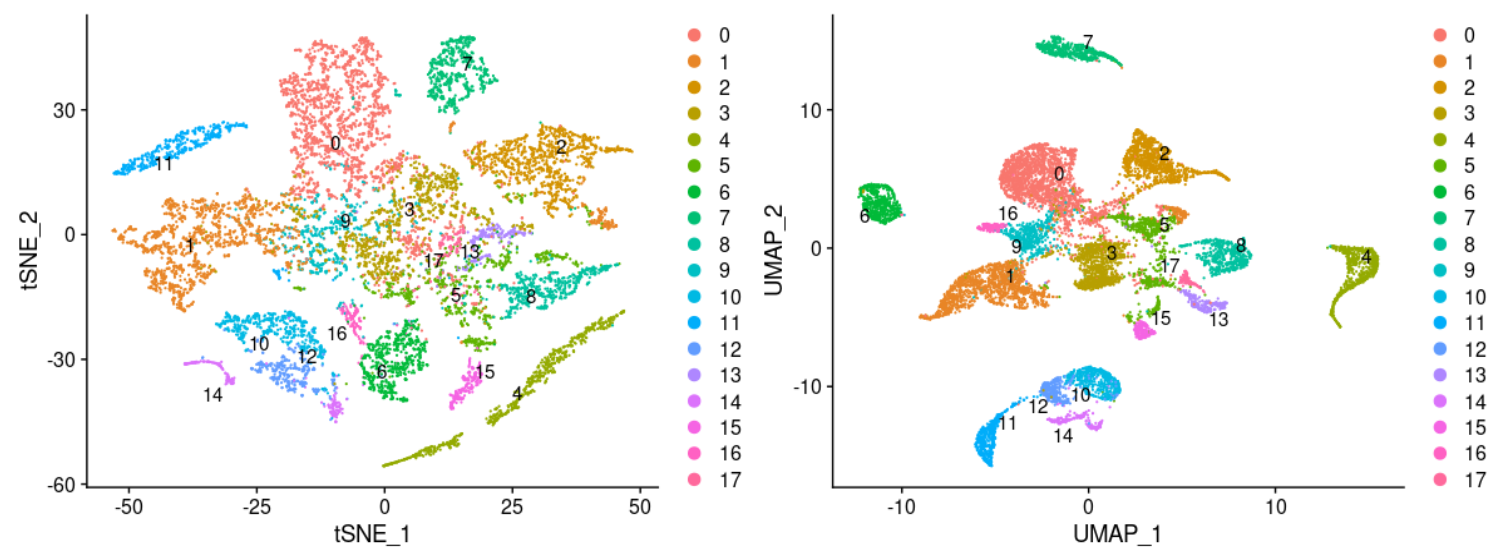
这个亚群数量使用的是resolution = 0.2,得到但是18个,其实并不好,因为癌症种类就有33个,理论上需要加大resolution ,得到更多的亚群哦!
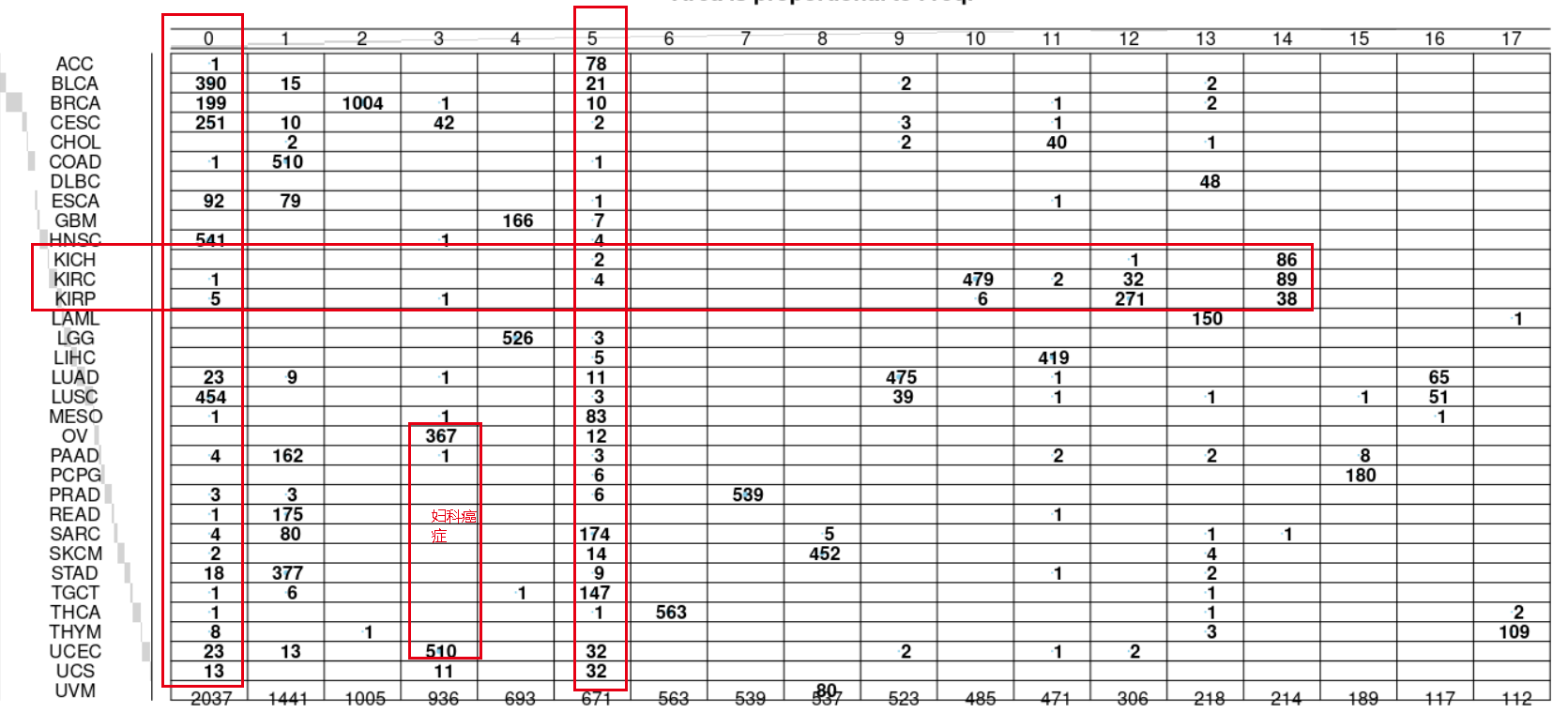
可以看到肾癌仍然是很有意思,妇科癌症OV和UCEC类似,在seurat的第0群里面绝大部分都是SC就是鳞癌,主要是LUSC,HNSC,CESC 等等。
因为这个时候的分辨率不够,可以看到 THCA 这个癌症的五百多个样品这个时候并没有被拆分成为两个亚群。感兴趣的小伙伴可以自己去测试看看:大resolution ,得到更多的亚群,看看这个时候是不是THCA就会被拆分!
对seurat默认的分群进行差异分析
这里我们仍然是使用 resolution = 0.2得到的亚群进行 FindAllMarkers函数处理,代码如下:
# 接下来对seurat默认的分群进行差异分析
pro='basic_seurat'
table(Idents(sce))
sce.markers <- FindAllMarkers(object = sce, only.pos = TRUE, min.pct = 0.25,
thresh.use = 0.25)
DT::datatable(sce.markers)
write.csv(sce.markers,file=paste0(pro,'_sce.markers.csv'))
library(dplyr)
top10 <- sce.markers %>% group_by(cluster) %>% top_n(10, avg_log2FC)
DoHeatmap(sce,top10$gene,size=3)
ggsave(filename=paste0(pro,'_sce.markers_heatmap.pdf'),width = 18,height = 21)
p <- DotPlot(sce, features = unique(top10$gene),
assay='RNA' ) + coord_flip()
p
ggsave(paste0(pro,'-DotPlot_check_top10_markers_by_clusters.pdf'),width = 18,height = 21)
library(dplyr)
top3 <- sce.markers %>% group_by(cluster) %>% top_n(3, avg_log2FC)
DoHeatmap(sce,top3$gene,size=3)
ggsave(paste0(pro,'-DoHeatmap_check_top3_markers_by_clusters.pdf'),width = 18,height = 11)
p <- DotPlot(sce, features = unique(top3$gene),
assay='RNA' ) + coord_flip()
p
ggsave(paste0(pro,'-DotPlot_check_top3_markers_by_clusters.pdf'),width = 18,height = 11)
save(sce.markers,file=paste0(pro,file = '-sce.markers.Rdata'))
前面我们提到了,在seurat的第0群里面绝大部分都是SC就是鳞癌,主要是LUSC,HNSC,CESC 等等。现在可以看第0群高表达量基因啦:
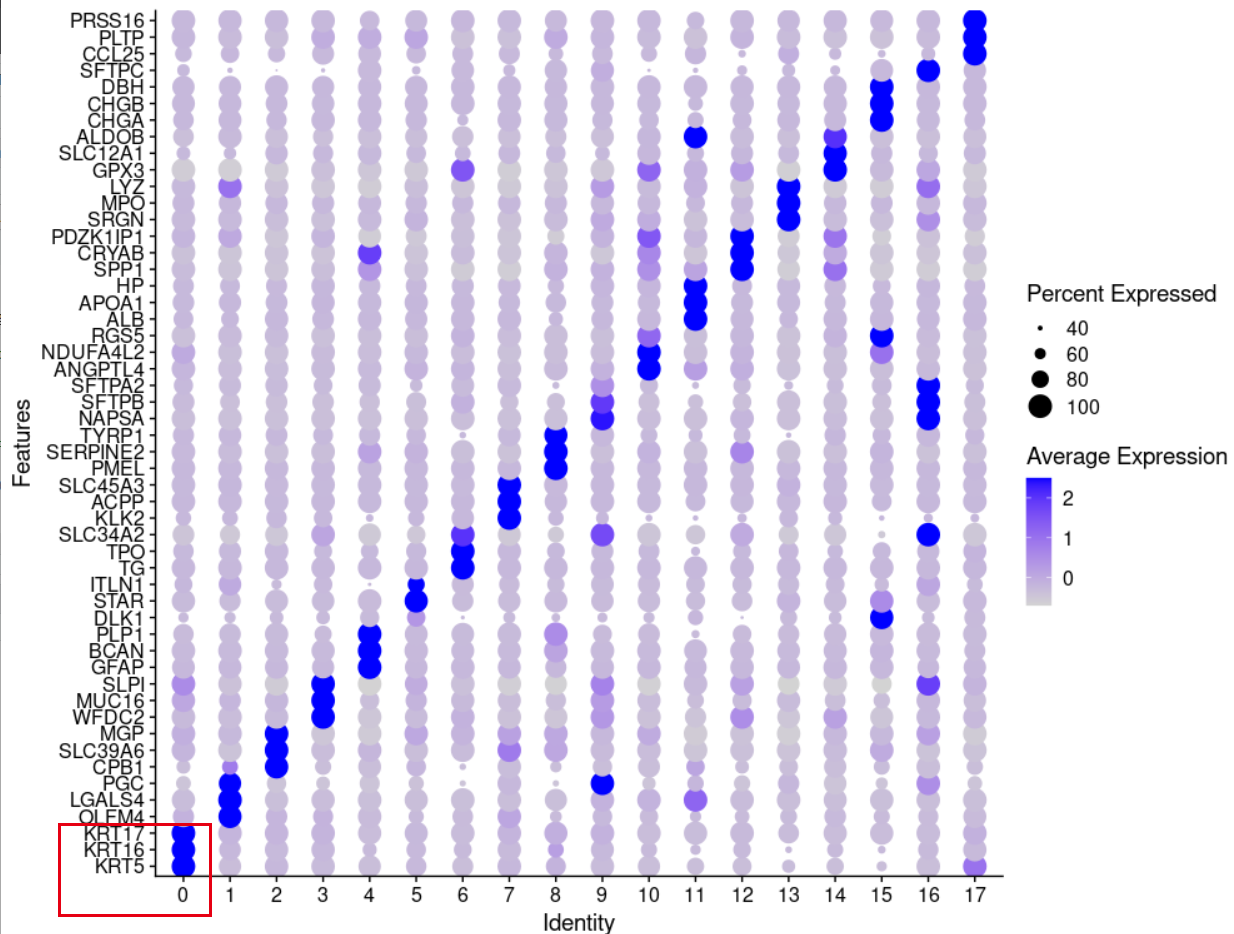
那问题来说,鳞癌为什么统一的高表达KRT16和KRT5呢?这是不是一个很好的课题呢?
同时保留这个单细胞的 seurat对象哦,后续我们的很多分析也是基于它 :
save(sce,file = 'sce.Rdata')
泛癌分析,就是如此简单!
单细胞数据看起来种类很多,有CEL-seq、MARS-seq、Drop-seq、Chromium 10x和SMART-seq的fastq数据。但是最终都是得到表达量矩阵哦, 大家通常是5个R包,分别是: scater,monocle,Seurat,scran,M3Drop,需要熟练掌握它们的对象,:一些单细胞转录组R包的对象 而且分析流程也大同小异:
- step1: 创建对象
- step2: 质量控制
- step3: 表达量的标准化和归一化
- step4: 去除干扰因素(多个样本整合)
- step5: 判断重要的基因
- step6: 多种降维算法
- step7: 可视化降维结果
- step8: 多种聚类算法
- step9: 聚类后找每个细胞亚群的标志基因
- step10: 继续分类
如果你也对10x单细胞转录组感兴趣,参考我们的《明码标价》专栏里面的单细胞内容
单细胞转录组数据分析的标准降维聚类分群,并且进行生物学注释后的结果。可以参考前面的例子:人人都能学会的单细胞聚类分群注释 ,我们演示了第一层次的分群。
如果你对单细胞数据分析还没有基础认知,可以看基础10讲: