一般来说单细胞表达量矩阵都是以基因为单位,我们可以很容易走常规的降维聚类分群并且合理的进行生物学命名,比如我们对官方 pbmc3k 例子,跑标准代码:
```r
library(Seurat)
# https://cf.10xgenomics.com/samples/cell/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz
## Load the PBMC dataset
pbmc.data <- Read10X(data.dir = "filtered_gene_bc_matrices/hg19/")
## Initialize the Seurat object with the raw (non-normalized data).
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k",
min.cells = 3, min.features = 200)
## Identification of mithocondrial genes
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
## Filtering cells following standard QC criteria.
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 &
percent.mt < 5)
## Normalizing the data
pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize",
scale.factor = 10000)
pbmc <- NormalizeData(pbmc)
## Identify the 2000 most highly variable genes
pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)
## In addition we scale the data
all.genes <- rownames(pbmc)
pbmc <- ScaleData(pbmc, features = all.genes)
pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc),
verbose = FALSE)
pbmc <- FindNeighbors(pbmc, dims = 1:10, verbose = FALSE)
pbmc <- FindClusters(pbmc, resolution = 0.5, verbose = FALSE)
pbmc <- RunUMAP(pbmc, dims = 1:10, umap.method = "uwot", metric = "cosine")
table(pbmc$seurat_clusters)
# pbmc.markers <- FindAllMarkers(pbmc, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25, verbose = FALSE)
DimPlot(pbmc, reduction = "umap", group.by = 'seurat_clusters',
label = TRUE, pt.size = 0.5)
DotPlot(pbmc, features = c("MS4A1", "GNLY", "CD3E",
"CD14", "FCER1A", "FCGR3A",
"LYZ", "PPBP", "CD8A"),
group.by = 'seurat_clusters')
## Assigning cell type identity to clusters
new.cluster.ids <- c("Naive CD4 T", "CD14+ Mono", "Memory CD4 T", "B", "CD8 T",
"FCGR3A+ Mono", "NK", "DC", "Platelet")
names(new.cluster.ids) <- levels(pbmc)
pbmc <- RenameIdents(pbmc, new.cluster.ids)
DimPlot(pbmc, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()
pbmc$cluster_by_counts=Idents(pbmc)
table(pbmc$cluster_by_counts)
```
得到的结果如下所示:
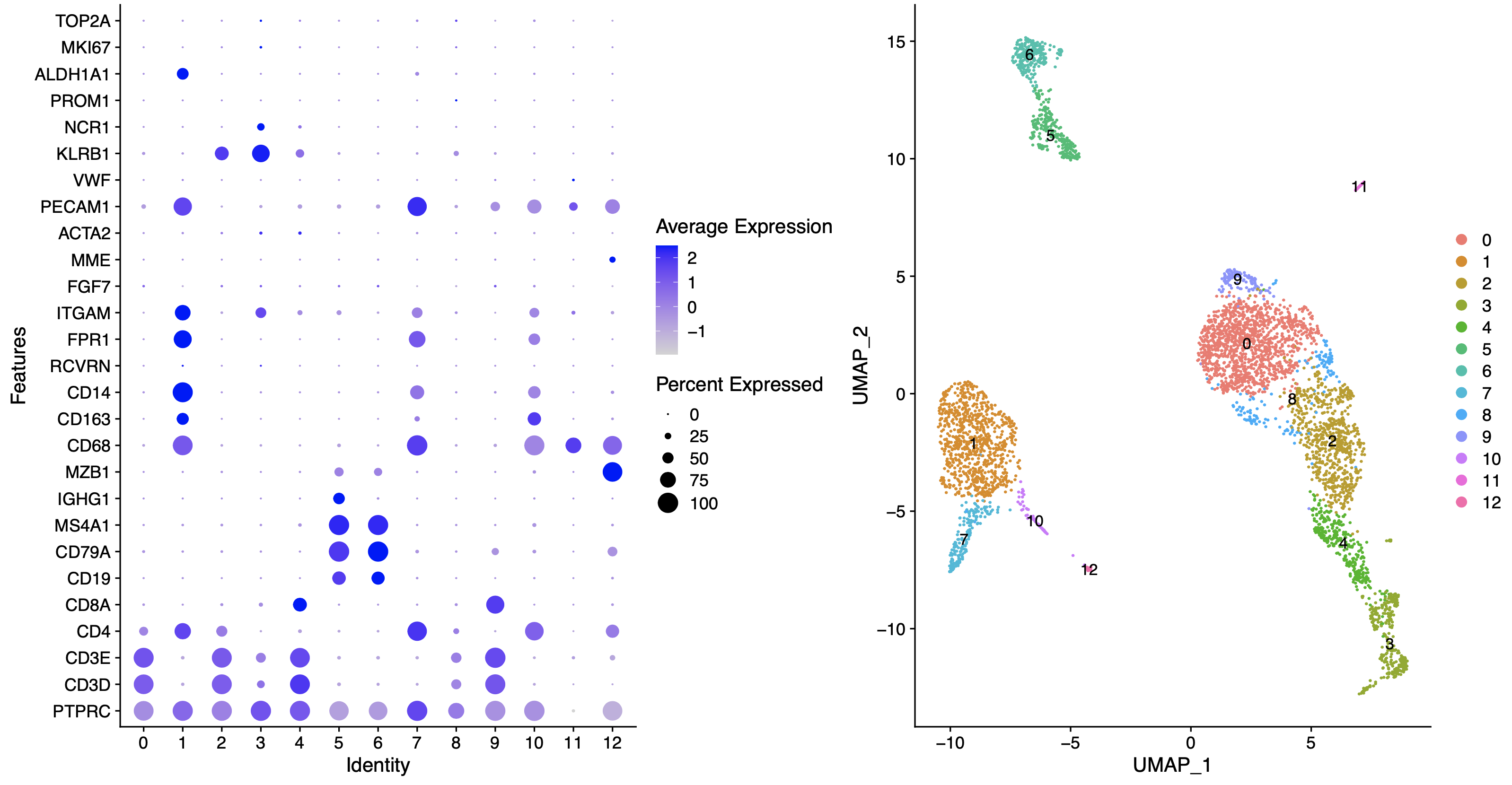
根据我们的**生物学背景**,可以进行如下所示的命名:
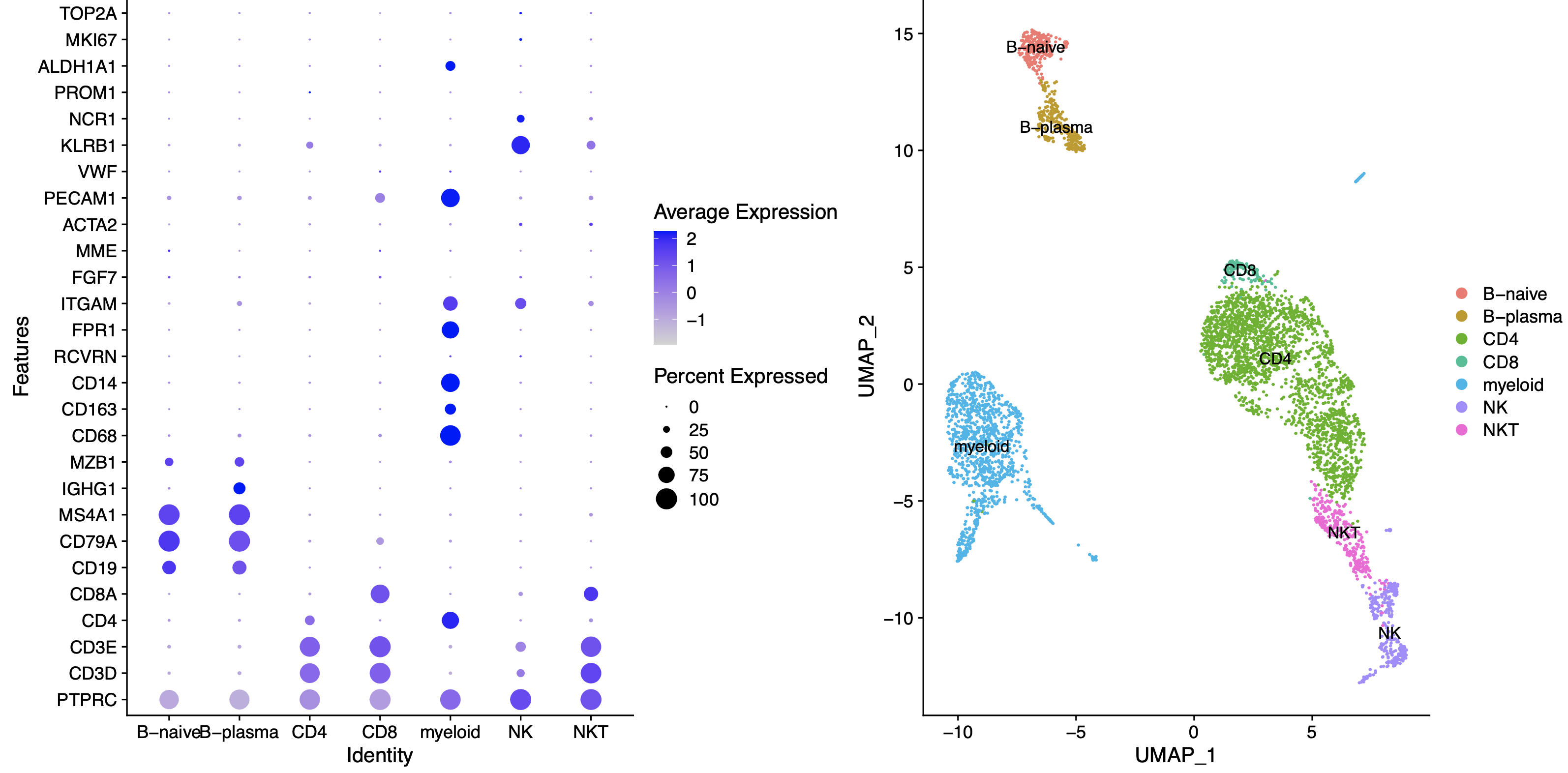
这个时候的生物学背景因人而异哦,比如我这里对myeloid了解不多,所以就没有细分出来巨噬细胞,单核细胞,树突细胞等等。仅仅是举例而已。
### 首先可以替换成为转录因子活性
这里推荐一个 R包,DoRothEA on http://bioconductor.org/ and https://github.com/saezlab/dorothea.
#### 获取包自带数据库
```r
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("dorothea")
library(tidyverse)
## We read Dorothea Regulons for Human:
dorothea_regulon_human <- get(data("dorothea_hs", package = "dorothea"))
## We obtain the regulons based on interactions with confidence level A, B and C
regulon <- dorothea_regulon_human %>%
dplyr::filter(confidence %in% c("A","B","C"))
```
#### 使用数据库去分析pbmc
```r
## We compute Viper Scores
library(dorothea)
pbmc <- run_viper(pbmc, regulon,
options = list(method = "scale", minsize = 4,
eset.filter = FALSE, cores = 1,
verbose = FALSE))
```
重新进行降维聚类分群:
```r
## We compute the Nearest Neighbours to perform cluster
DefaultAssay(object = pbmc) <- "dorothea"
pbmc <- ScaleData(pbmc)
pbmc <- RunPCA(pbmc, features = rownames(pbmc), verbose = FALSE)
pbmc <- FindNeighbors(pbmc, dims = 1:10, verbose = FALSE)
pbmc <- FindClusters(pbmc, resolution = 0.5, verbose = FALSE)
pbmc <- RunUMAP(pbmc, dims = 1:10, umap.method = "uwot", metric = "cosine")
DimPlot(pbmc, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()
library(gplots)
balloonplot(table(pbmc$dorothea_snn_res.0.5,
pbmc$cluster_by_counts))
```
同一个10x单细胞数据的两个矩阵各自的降维聚类分群是类似的,但是又不能完全一致:
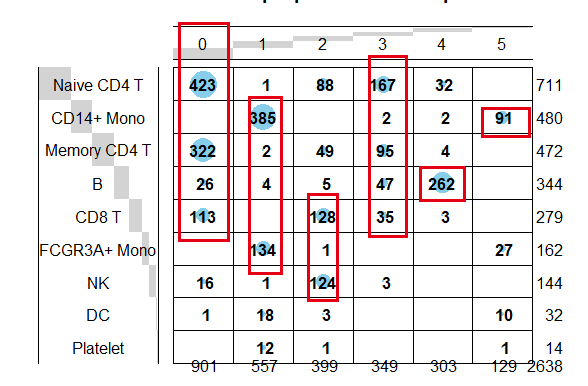
这个时候的表达量矩阵并不是原始的每个基因在每个细胞里面的counts值了哦,而是每个转录因子在每个细胞的活性情况,我们也可以人工命名,
后面也可以对这样的矩阵(每个转录因子在每个细胞的活性情况) ,进行差异分析等等,参考;https://bioconductor.org/packages/release/data/experiment/vignettes/dorothea/inst/doc/single_cell_vignette.html
### 也可以可以替换成为gsea打分
单位就是各个基因集,比如MSigDB(Molecular Signatures Database)数据库中定义了已知的基因集合:http://software.broadinstitute.org/gsea/msigdb 包括H和C1-C7八个系列(Collection),每个系列分别是:
- H: hallmark gene sets (癌症)特征基因集合,共50组,最常用;
- C1: positional gene sets 位置基因集合,根据染色体位置,共326个,用的很少;
- C2: curated gene sets:(专家)校验基因集合,基于通路、文献等:
- C3: motif gene sets:模式基因集合,主要包括microRNA和转录因子靶基因两部分
- C4: computational gene sets:计算基因集合,通过挖掘癌症相关芯片数据定义的基因集合;
- C5: GO gene sets:Gene Ontology 基因本体论,包括BP(生物学过程biological process,细胞原件cellular component和分子功能molecular function三部分)
- C6: oncogenic signatures:癌症特征基因集合,大部分来源于NCBI GEO 发表芯片数据
- C7: immunologic signatures: 免疫相关基因集合。
全新的矩阵是基因集在每个细胞的打分矩阵,然后可以再次聚类看看!
### 作为学徒作业
大家需要拿kegg的约300个通路,对这个pbmc3k单细胞表达量矩阵进行ssGSEA打分后得到矩阵,进行降维聚类分群!
详情:https://mp.weixin.qq.com/s/-jAaHAvraGnVLsPLGKTvQA