太多人咨询基因的各种ID转换问题,在非模式生物的物种里面更麻烦,因为数据库注释资源并不权威。
但是实际上这样的基因ID转换也不是必须的,因为差异分析要的是表达量矩阵,基因名字并不重要啊,后面的注释也是可以基于ID,一步到位成为功能。比如文章:The effects of Arabidopsis genome duplication on the chromatin organization and transcriptional regulation. Nucleic Acids Res 2019 Sep 5;47(15):7857-7869. PMID: 31184697
数据链接是:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE114950
可以看到里面的转录组测序样品是2个分组, 每个组内是3个重复,共6个样品:
GSM3529059 rna_seq_col-rep1_RRAS07926-V
GSM3529060 rna_seq_col-rep2_RRAS07927-V
GSM3529061 rna_seq_col-rep3_RRAS07928-V
GSM3529062 rna_seq_s4XCol-rep1_RRAS07929-V
GSM3529063 rna_seq_s4XCol-rep2_RRAS07930-V
GSM3529064 rna_seq_s4XCol-rep3_RRAS07931-V
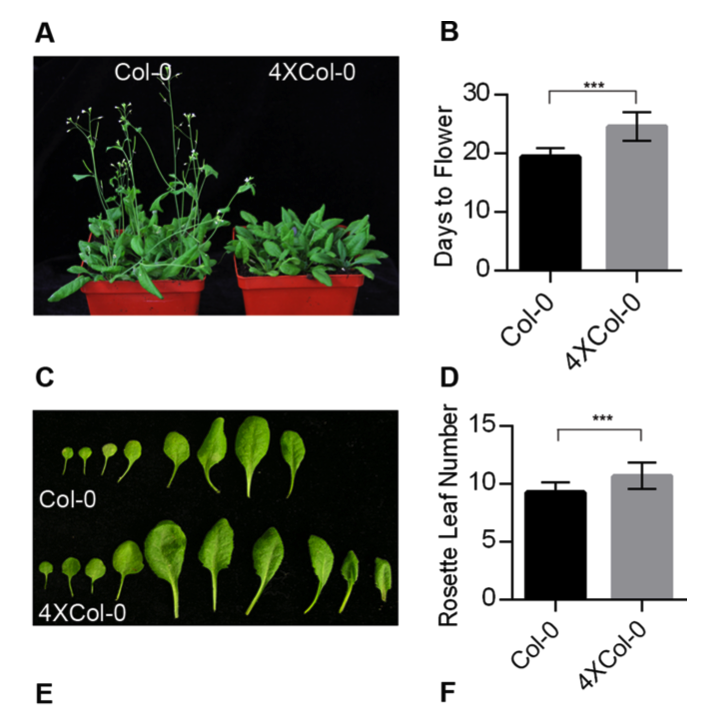
这两个分组是有形态学差异的, 如下所示: Col-0 and autotetraploid (4 × Col-0) Arabidopsis.,所以做全局的转录组数据,去探索这两个分组的差异基因及其功能。
转录组的标准分析,比较容易复现,基本上看我六年前的表达芯片的公共数据库挖掘系列推文即可;
- 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够
如下所示展示了上下调基因的火山图:
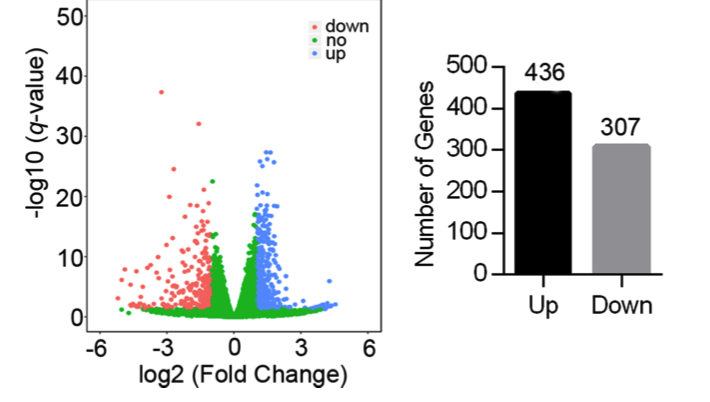
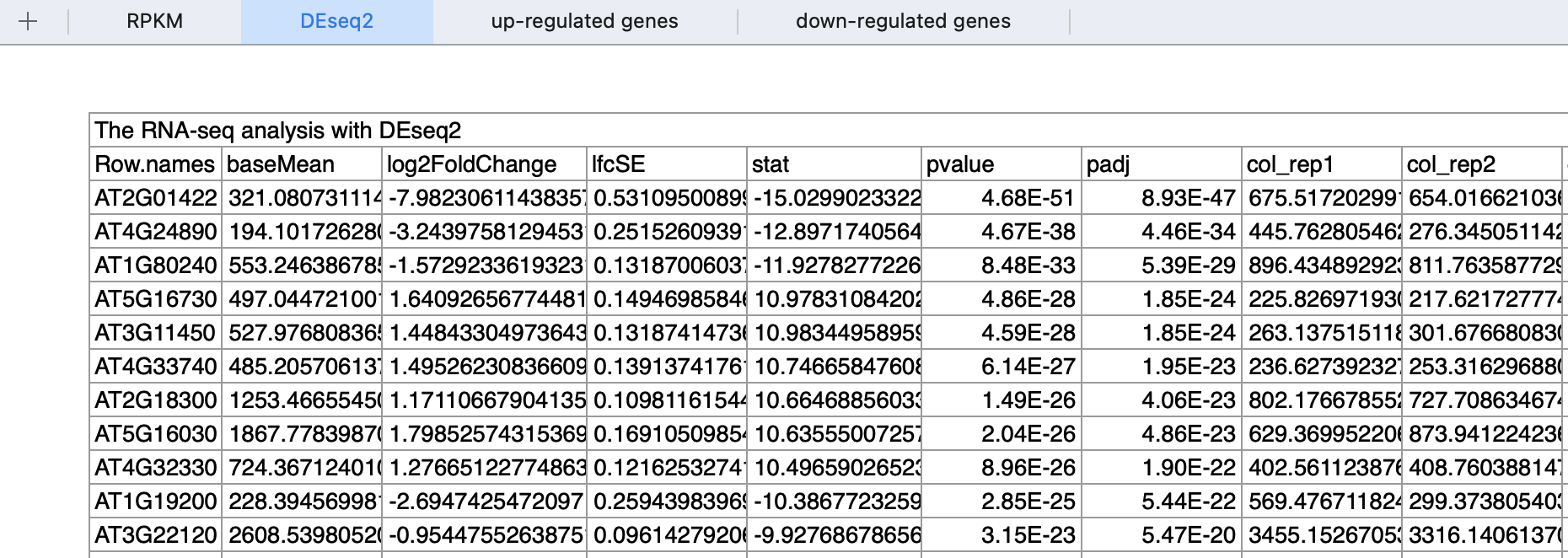
这里的阈值是 (|log2fold change| > 1) ,采用DEseq2这个方法,在附件也给出来了差异分析结果:
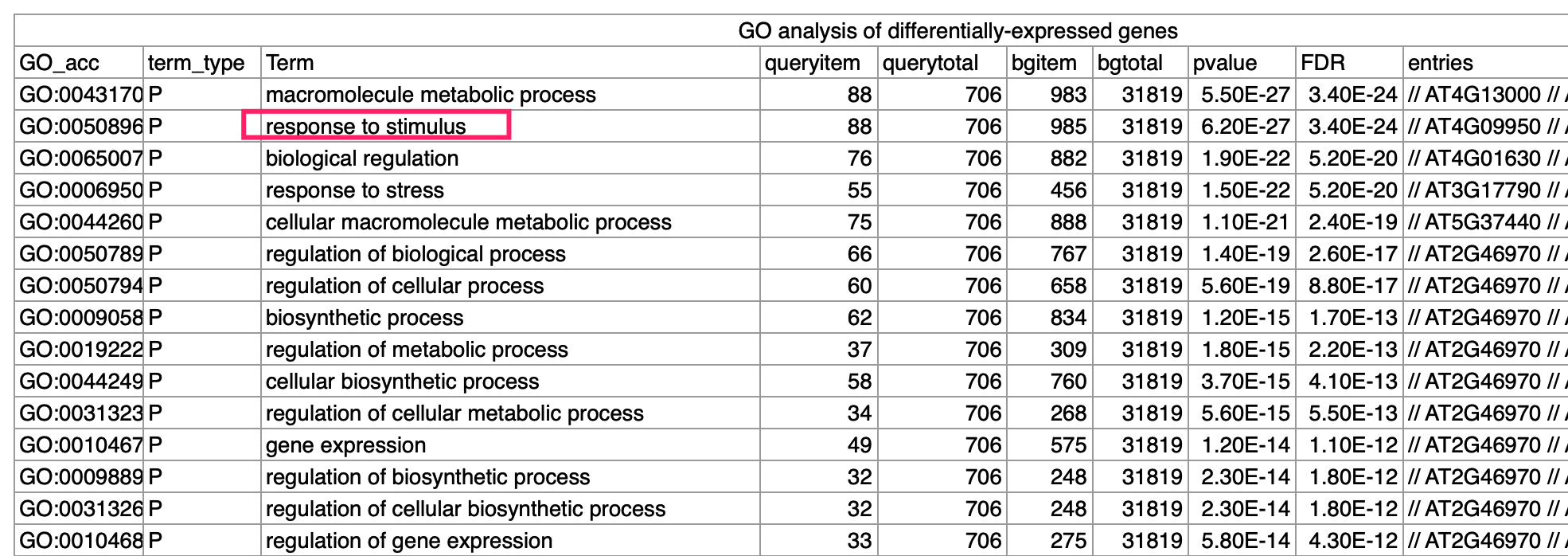
但是接下来需要看上下调基因的功能,这个时候基因的名字仍然是可以不需要,使用基因的一个ID即可,比如文章做的是GO数据库富集分析注释 :
Gene ontology (GO) analysis revealed that these differen- tially expressed genes associate with responses to starvation, metabolic process and stimuli ,在附件也给出来了富集分析结果:
差异分析的结果和富集分析的结果都是在附表: - Supplementary Table 1. List of the differentially expressed genes between Col-0 and 4 ×Col-0 Arabidopsis.
- Supplementary Table 2. GO terms of the differentially expressed genes in 4×Col-0 compared to Col-0.
也就是说, 只要你有一个全面打通的命名系统,它未必需要符合人类的认知,尽管是AT开头的基因ID,人类是不太可能一眼看出来它背后的意义。但是计算机可以,计算机可以高速查询。
那么问题来了,基因名字在什么时候需要使用呢?比如在单细胞亚群命名:
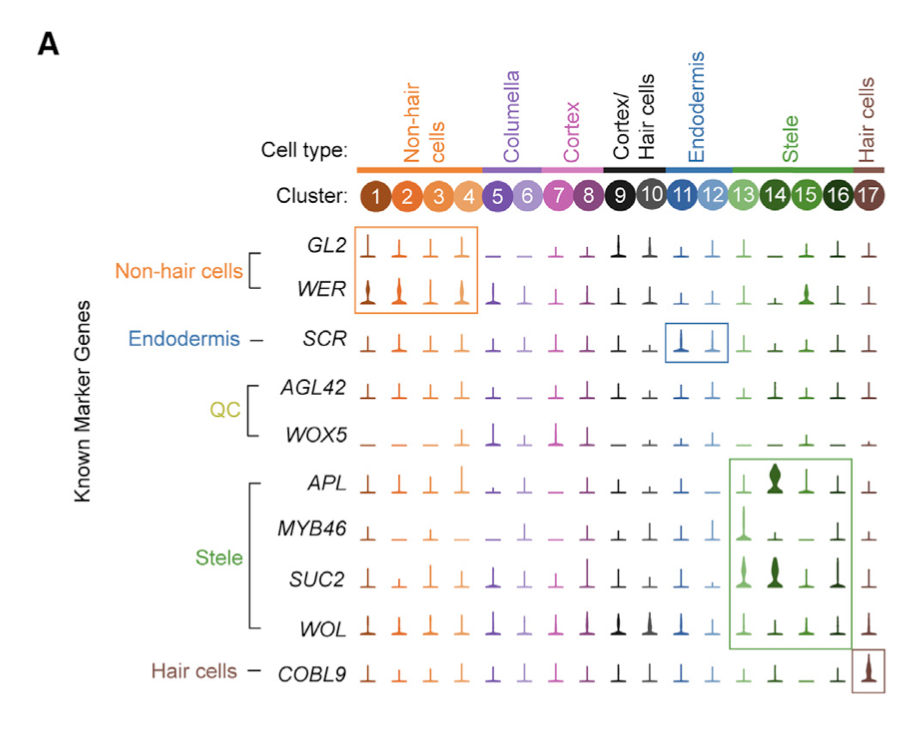
文章是《High-Throughput Single-Cell Transcriptome Profiling of Plant Cell Types》,里面的图(A) Violin plots showing the expression of 10 commonly used cell type marker genes across all clusters.
这个时候就需要做基因ID转换了,拟南芥的基因ID转换也无非是找到一个基因对应表格而已!