众所周知10x单细胞会给出3个文件,我在单细胞数据分析的基础10讲写的很清楚:
- 01. 上游分析流程
- 02.课题多少个样品,测序数据量如何
- 03. 过滤不合格细胞和基因(数据质控很重要)
- 04. 过滤线粒体核糖体基因
- 05. 去除细胞效应和基因效应
- 06.单细胞转录组数据的降维聚类分群
- 07.单细胞转录组数据处理之细胞亚群注释
- 08.把拿到的亚群进行更细致的分群
- 09.单细胞转录组数据处理之细胞亚群比例比较
如果是10x的单细胞公共数据,比如 GSE128033 和 GSE135893,就是10x数据集,随便下载其中一个,就能看到每个样本都是走流程拿到10x单细胞转录组数据的3个文件的表达矩阵。
2.2M Mar 8 2019 GSM3660655_SC94IPFUP_barcodes.tsv.gz
259K Mar 8 2019 GSM3660655_SC94IPFUP_genes.tsv.gz
26M Mar 8 2019 GSM3660655_SC94IPFUP_matrix.mtx.gz
2.2M Mar 8 2019 GSM3660656_SC95IPFLOW_barcodes.tsv.gz
259K Mar 8 2019 GSM3660656_SC95IPFLOW_genes.tsv.gz
31M Mar 8 2019 GSM3660656_SC95IPFLOW_matrix.mtx.gz
2.2M Mar 8 2019 GSM3660657_SC153IPFLOW_barcodes.tsv.gz
259K Mar 8 2019 GSM3660657_SC153IPFLOW_genes.tsv.gz
33M Mar 8 2019 GSM3660657_SC153IPFLOW_matrix.mtx.gz
2.2M Mar 8 2019 GSM3660658_SC154IPFUP_barcodes.tsv.gz
259K Mar 8 2019 GSM3660658_SC154IPFUP_genes.tsv.gz
31M Mar 8 2019 GSM3660658_SC154IPFUP_matrix.mtx.gz
每个10x样品,都是3个文件,规律很明显,下游处理的时候,一定要保证这3个文件同时存在,而且在同一个文件夹下面。
示例代码是:
rm(list=ls())
options(stringsAsFactors = F)
library(Seurat)
sce1 <- CreateSeuratObject(Read10X('../10x-results/WT/'),
"wt")
重点就是 Read10X 函数读取 文件夹路径,比如:../10x-results/WT/ ,保证文件夹下面有3个文件,而且文件名只能是 barcodes.tsv.gz 和 matrix.mtx.gz 以及 genes.tsv.gz,绝对是不能被混杂的 。
但是并不是所有的文献在共享数据的时候都是那么标准,比如文章:https://science.sciencemag.org/content/suppl/2018/08/13/361.6402.594.DC1
作者在附件,给出来的就是一个纯粹的 矩阵文件,后缀是 mtx,然后配合了两个tsv文件,并不是标准的10x的3个文件!
A table of counts for all candidate droplets (i.e., the 125,139 unique barcodes at the top of Fig. S1). Stored in matrix mart format along with two extra tables describing the column labels (droplets) and row labels (genes). Counts represent number of unique UMIs for each gene/droplet pair (see Methods).
然后需要解压:
然后进入直接读取:
library(Seurat)
# https://hbctraining.github.io/scRNA-seq/lessons/readMM_loadData.html
library(Matrix)
mtx=readMM('tableOfCounts.mtx')
mtx
dim(mtx)
cl=read.table('tableOfCounts_colLabels.tsv',
header = T)[,2]
rl=read.table('tableOfCounts_rowLabels.tsv',
header = T)[,2]
head(cl)
head(rl)
rownames(mtx) <- rl
colnames(mtx) <- cl
sce=CreateSeuratObject(counts = mtx)
可以看到样本名以及基因名字都还是蛮复杂的:
> head(cl)
[1] "4602STDY7018923___AAACGGGGTCCAACTA"
[2] "4602STDY7018923___AAAGATGAGGAGTACC"
[3] "4602STDY7018923___AAAGATGGTGAACCTT"
[4] "4602STDY7018923___AAAGTAGTCCCTTGTG"
[5] "4602STDY7018923___AAATGCCCAGTAAGAT"
[6] "4602STDY7018923___AACCATGCAGTCGTGC"
> head(rl)
[1] "RP11-34P13.3_ENSG00000243485" "FAM138A_ENSG00000237613"
[3] "OR4F5_ENSG00000186092" "RP11-34P13.7_ENSG00000238009"
[5] "RP11-34P13.8_ENSG00000239945" "RP11-34P13.14_ENSG00000239906"
后续可以慢慢修饰这个sce对象啦!也就是说 readMM 函数即可,然后配合CreateSeuratObject来构建对象!
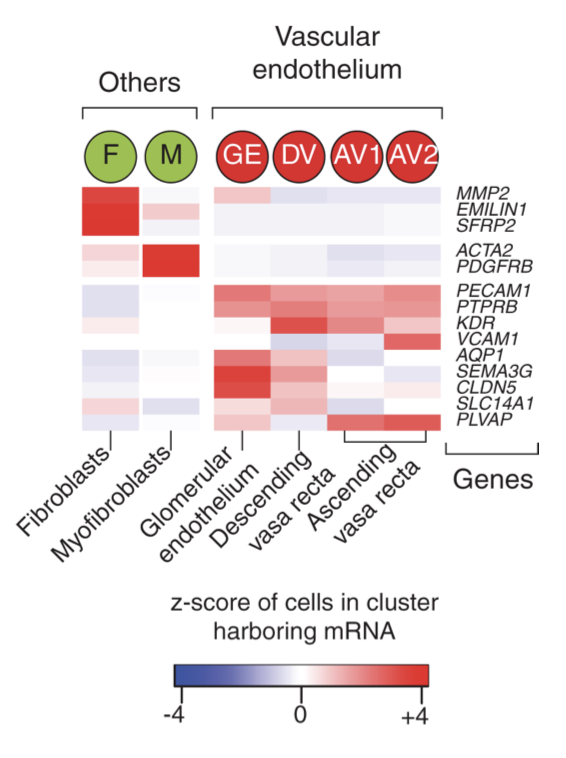
降维聚类分群和生物学注释都走起!不过,因为这个文章是早期单细胞,所以数据质量并不是太好,分析的过程可能会遇到意外:
当然了,如果你是10x的fastq文件,需要10X单细胞转录组数据的cellranger结果文件,我们也是在单细胞天地公众号详细介绍了cellranger全部使用细节及流程,大家可以自行前往学习,如下:
- 单细胞实战(一)数据下载
- 单细胞实战(二) cell ranger使用前注意事项
- 单细胞实战(三) Cell Ranger使用初探
- 单细胞实战(四) Cell Ranger流程概览
- 单细胞实战(五) 理解cellranger count的结果
- 更新版本见:cellranger更新到4啦(全新使用教程)
因为这个cellranger上游数据分析流程对服务器要求比较高,而且10x的原始测序fastq文件也是接近100G文件,运行过程对技术资源消耗比较大。