如果你手上有一套尘封已久的转录组表达量芯片数据或者测序数据,那么这个文章或许是一个较好的模范让你学习!
文章标题是:《Systematic Identification of Hub Genes in Placenta Accreta Spectrum Based on Integrated Transcriptomic and Proteomic Analysis》,链接是:https://www.frontiersin.org/articles/10.3389/fgene.2020.551495/full 真的是超级简单的实验设计啦,关心的是placenta accreta spectrum (PAS),这个疾病,然后样品是 5 patients with PAS and 5 healthy pregnant women. 超级简介的实验设计,数据也是公开可以获取的!
- The BioProject Number is PRJNA627183 and you can learn more about the data at http://www.ncbi.nlm.nih.gov/bioproject/627183.
上游分析拿到这个转录组测序数据的表达量矩阵,大家应该是不会陌生了,转录组的流程我们其实多次反复分享了:视频观看方式 - 视频免费在B站:https://www.bilibili.com/video/BV12s41137HY 大家学习的时候记得发弹幕交流哈。
- 也有微云离线版本视频下载本地播放:
-
- 上游分析视频以及代码资料在:https://share.weiyun.com/5QwKGxi
- 下游主要是基于counts矩阵的标准分析的代码 https://share.weiyun.com/50hfuLi
- RNA-SEQ实战演练的素材:https://share.weiyun.com/5h1Z2QY ,包括一些公司PPT,综述以及文献以及测试数据
- RNA-SEQ 实战演练的思维导图:文档链接:https://mubu.com/doc/38y7pmgzLg 密码:p6fo
感兴趣的小伙伴,如果有自己的服务器,马上就可以抽几分钟,下载这个数据,去走这个流程,分享这个表达量矩阵给大家,欢迎在公众号这个推文下面留言交流哦!最简单的当然是差异分析
差异分析相信大家都不陌生了,基本上看我六年前的表达芯片的公共数据库挖掘系列推文即可;
- 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够
差异分析的前提是数据合理,我们对转录组会要求相关性图,主成分分析图,带有层次聚类的热图来说明组间差异是大于组内差异的。如下所示,就是一个表达量全局相关性,可以看到,同一个组相关性要远大于与其它组的:
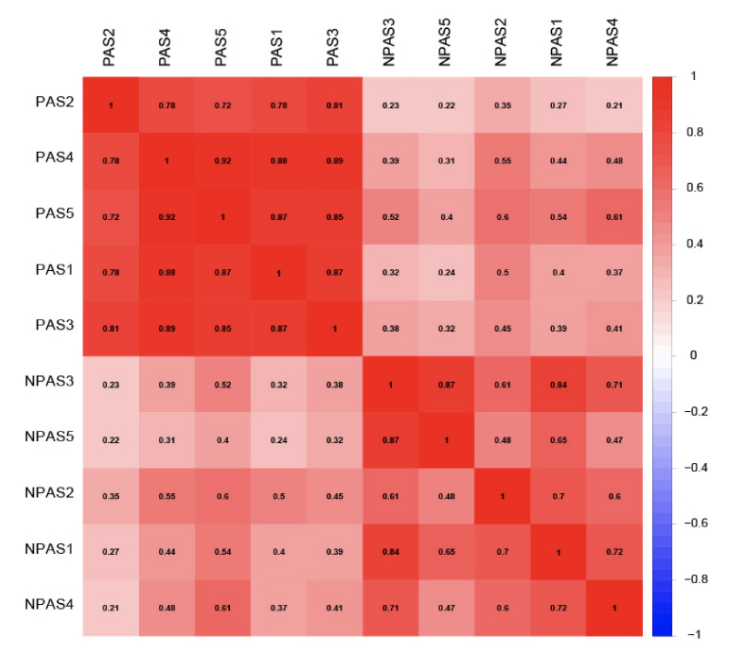
作者这里使用了 edgeR,结果如下: - A total of 17,860 known mRNAs in placenta tissue were detected quantitatively.
- And 728 differentially expressed mRNAs (false discovery rate, FDR < 0.05, fold change >1.5) were obtained,
- including 481 up-regulated genes and 247 down-regulated genes
一般来说,会以火山图的形式展现这个差异分析结果:
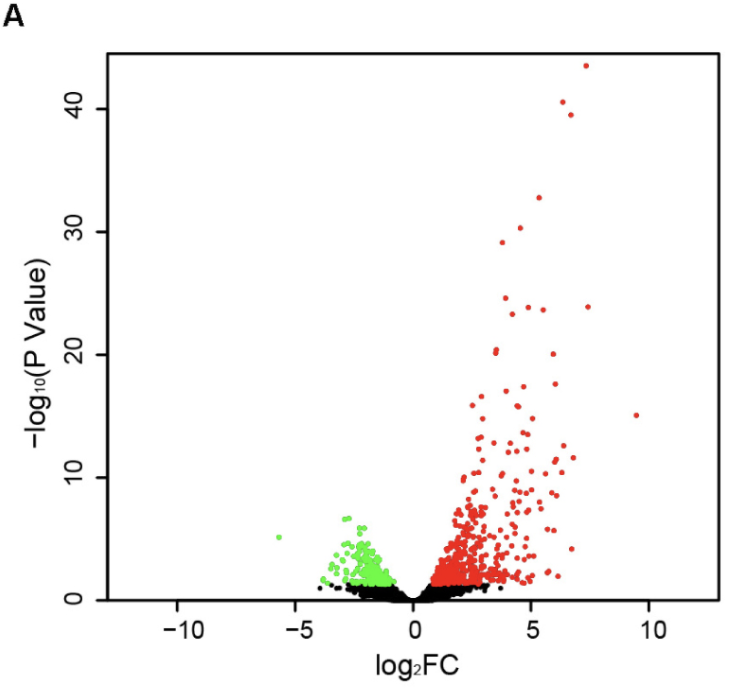
到这里,你的分析仅仅是一个公司的标准服务罢了,距离发表路还很长。接下来常规就是kegg和go数据库的注释啦,说明那些上下调的几百个基因的生物学功能。差异基因列表找hub基因
有了差异基因,另外一个主线任务就是拿到基因对应的蛋白之间的相互作用,使用 MCODE in Cytoscape . 就可以挑选多个不同的子网络关系, 各自寻找hub基因啦!
这个操作,我们有一个公开课,在:“cytoscape网络图绘制”群的钉钉群号: 34970303

关键是其实这个文章不仅仅是标准分析,他把转录组数据基本上能做的都做了个遍!转录组更多分析
当然了,转录组肯定不只是表达量这一个分析点,这个文章就做了融合基因的检查以及变异位点查询,还有可变剪切,软件列表如下:
- Soapfuse (version 1.18) (Jia et al., 2013) was used to detect the fusion gene in each sample.
- The Genome Analysis Toolkit (GATK, version 3.4-0) (McKenna et al., 2010) was used to detect single nucleotide polymorphisms (SNP) information in each sample,
- which was further annotated by Snpeff (Cingolani et al., 2012).
- ASprofile2 was used to quantitatively detect splicing events of each sample.
基本上都是描述性结果罗列即可: - Figure 7. Waterfall plot demonstrates the types of SNP in hub genes.
- Figure 8. Alternative splicing events of (A) all identified genes and (B) hub genes.
- Figure 9. Gene fusion between INHA and STK11IP in (A) PAS1, (B) PAS2, and (C) PAS5.
这些分析有很多难度也不小哦,如果大家完成起来有困难,可以考虑我们的《明码标价》服务,涵盖的数据分析包括但是不限于: - ATAC-seq项目的标准分析仅收费1600
- 单细胞转录组的质控降维聚类分群和生物学注释仅收费800
- 普通转录组上游分析仅收费800
- 公共数据库的WGCNA分析仅需800
- 公共数据库的生存分析进需800
- 明码标价之转录组常规测序服务(仅需799每个样品)
- 明码标价之转录组下游分析仅需800元
一个好玩的结论
这个研究居然还去对应的公共数据库的 GSE126552,然后发现这个GSE126552数据集居然是没有差异基因的,而且文章自己的这个PRJNA627183数据集分析拿到的hub基因没办法在这个公共数据集里面验证成功。
我就纳闷了,一个阴性结果他们怎么就这么大胆子写出来了呢?
Only one relevant sequencing data set can be found on Gene Expression Omnibus (GSE126552), in which PAS and normal placenta tissue was sequenced to find potential biomarkers and molecular mechanism of PAS. We downloaded and analyzed the data set, but unfortunately, there were no differentially expressed lncRNAs and mRNAs in this data set. Therefore, the hub genes found in this study cannot be effectively verified externally. - https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE126552
芯片平台是:Agilent-079487 Arraystar Human LncRNA microarray V4GSM3604340 Patient 1 Pc03p GSM3604341 Patient 2 Pc04p GSM3604342 Patient 3 Pc05p GSM3604343 Patient 4 Pc07p GSM3604344 Patient 5 Pc11p GSM3604345 Patient 1 Pp03p GSM3604346 Patient 2 Pp04p GSM3604347 Patient 3 Pp05p GSM3604348 Patient 4 Pp07p GSM3604349 Patient 5 Pp11p其实我怀疑可能是作者在分析这个公共数据库的 GSE126552可能是有失误,毕竟这个芯片平台是Agilent啊!!!