GTEx数据库想必大家并不陌生了,通常我们在挖掘TCGA数据库的时候,会发现该项目纳入的正常组织测序结果是非常少的,也就是说很多病人都不会有他的正常组织的转录组测序结果。
比如说乳腺癌吧,1200个左右的转录组数据,其中1100左右都是肿瘤组织的测序数据,只有区区100个左右的正常对照。这个时候我们就需要想办法加大正常组织测序样本量,既然TCGA数据库没有,我们就从其他数据库着手。这里值得大力推荐的是GTEx数据库 ,Genotype-Tissue Expression (GTEx)
最近有粉丝询问, 他想做小鼠的不同癌症模型,想问问看能不能纳入人类研究领域的GTEx数据库,如何去批次效应,如何转换基因名字。
我觉得这纯粹是绕远路了啊,简单搜索一下大鼠或者小鼠研究自己的数据库资源不是更好吗。
比如:于2017年12月发表在杂志的:An RNA-Seq atlas of gene expression in mouse and rat normal tissues
- 链接是:https://www.nature.com/articles/sdata2017185
- The dataset provides the transcriptome across tissues from three male C57BL6 mice and three male Han Wistar rats.
- 数据集上传到了:ArrayExpress E-MTAB-6081 (2017)
A complete list of the 77 tissue samples with sample ids is given in Table 1.
详细信息如下:
| Design Type(s) | species comparison design • organism part comparison design |
|---|---|
| Measurement Type(s) | transcription profiling assay |
| Technology Type(s) | RNA sequencing |
| Factor Type(s) | Species • animal body part |
| Sample Characteristic(s) | Mus musculus • Rattus norvegicus • brain • colon • duodenum • esophagus • heart • ileum • jejunum • kidney • liver • pancreas • quadriceps femoris • stomach • thymus |
看了看,走的是STAR aligner 流程,有意思的是在使用featureCounts定量之前居然对bam文件进行了去重操作。
因为是RNA-seq数据,所以提供了counts, rpkm, tpm这3种形式的数据
R-code provided in Supplementary S2. 感兴趣的可以看看:
## load the data (counts, rpkm, tpm)
mouseEnv$counts <- read.table("./data/mouse_counts.txt", check.names = F)
mouseEnv$rpkm <- read.table("./data/mouse_rpkm.txt", check.names = F)
mouseEnv$tpm <- read.table("./data/mouse_tpm.txt", check.names = F)
mouseEnv$pre.design <- read.table("./data/mouse_design.txt", check.names = F)
ratEnv$counts <- read.table("./data/rat_counts.txt", check.names = F)
ratEnv$rpkm <- read.table("./data/rat_rpkm.txt", check.names = F)
ratEnv$tpm <- read.table("./data/rat_tpm.txt", check.names = F)
ratEnv$pre.design <- read.table("./data/rat_design.txt", check.names = F)
研究者采用limm做差异分析
蛮简单的:
## use limma to get voom-normalized log(cpm) values
for (e in c(mouseEnv, ratEnv)) {
group <- factor(e$pre.design[,"group"])
design <- model.matrix(~0+group)
colnames(design) <- levels(group)
## put counts into DGE object and normalize
dge <- DGEList(counts=e$counts)
## ignore if not expressed in at least one sample
isexpr <- rowSums(cpm(dge)>1) >= 1
dge <- dge[isexpr,keep.lib.sizes=FALSE]
dge <- calcNormFactors(dge)
e$v <- voom(dge,design)
}
附件的R代码也可以学习下,比如下面这个图,难道你不想知道是如何绘制的吗?:
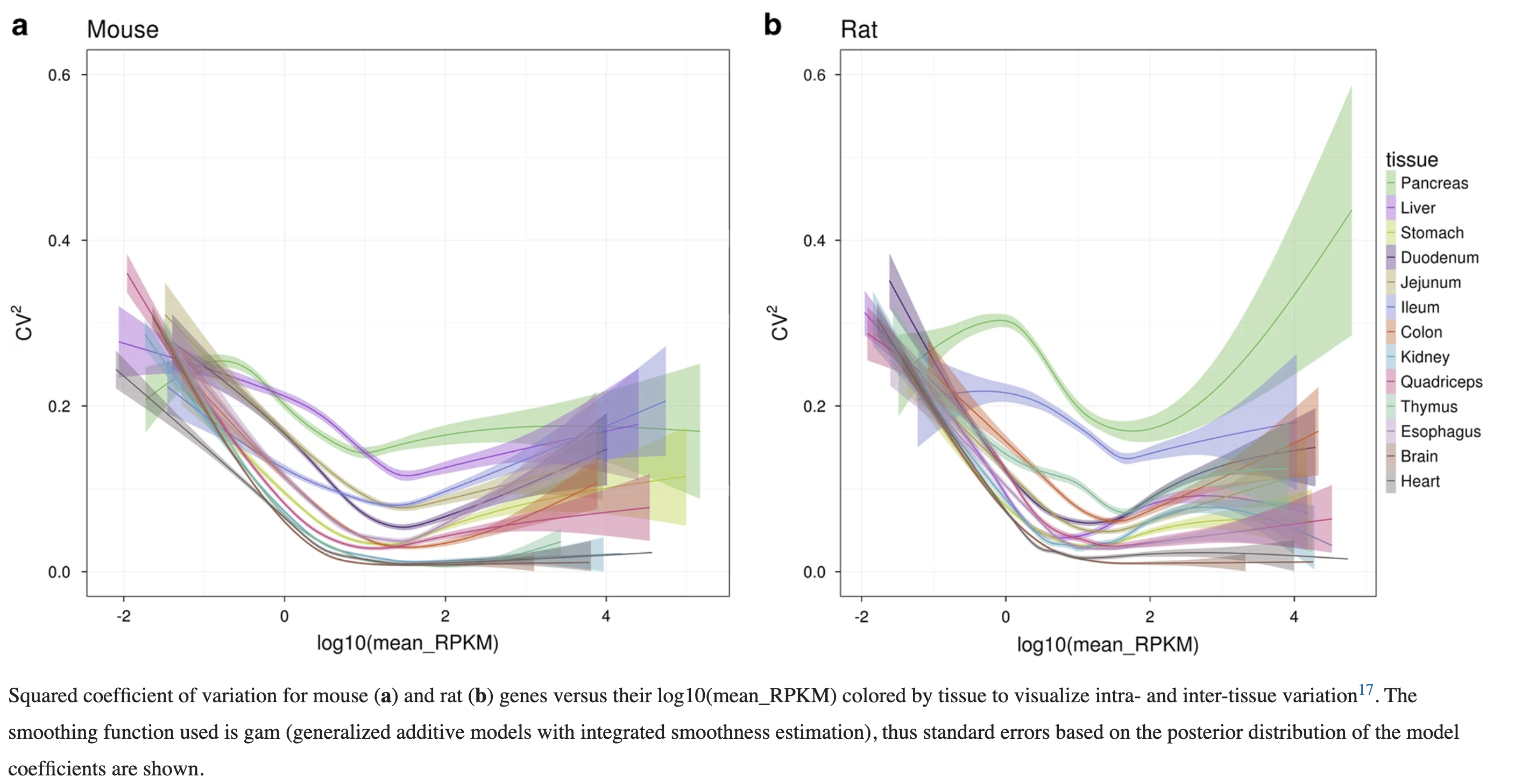
当然了,需要R语言基础,不然看起来磕磕碰碰。