最近有粉丝在我们《生信技能树》公众号后台吐槽说某公司,给他们测了ATAC-seq,只拿到差异peak,想要不差异的peak居然被告之是额外分析项目,要加钱。各种巧立名目的费用让他害怕,还不如直接找公司要来fastq测序数据,找我们从头开始分析。
一条龙服务,一个ATAC-seq项目的标准分析仅收费1600。同样的我把这个《ATAC-seq》任务安排给了学徒,感谢学徒在这个春节假期还兢兢业业完成任务!
下面是学徒的探索
环境搭建
如果是全新服务器或者全新用户,首先需要安装conda(最适合初学者的软件管理解决方案):
#一路yes下去
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
bash Miniconda3-4.6.14-Linux-x86_64.sh
source ~/.bashrc
然后使用conda安装一些软件或者软件环境,比如下载测序数据文件的aspera软件环境:
conda create -n download -y
conda activate download
conda install -y -c hcc aspera-cli
which ascp
## 一定要搞清楚你的软件被conda安装在哪
ls -lh ~/miniconda3/envs/download/etc/asperaweb_id_dsa.openssh
还有ATAC-SEQ数据分析流程的相关软件:
## 安装好conda后需要设置镜像。
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/conda-forge
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/bioconda
conda config --set show_channel_urls yes
conda create -n atac -y python=2 bwa
conda info --envs
# 可以用search先进行检索
conda search trim_galore
## 保证所有的软件都是安装在 atac 这个环境下面
conda activate atac
conda install -y trim-galore bedtools deeptools homer meme macs2 bowtie bowtie2 sambamba
conda search samtools
conda install -y samtools=1.11
然后构建工作目录架构:
# 注意组织好自己的项目
mkdir -p ~/project/atac/
cd ~/project/atac/
mkdir {sra,raw,clean,align,peaks,motif,qc}
cd raw
取决于个人习惯。
实战数据准备
参考:使用ebi数据库直接下载fastq测序数据 , 需要自行配置好,然后去EBI里面搜索到的 fq.txt 路径文件:
- 项目地址是
#一次性下载所有的 fastq.gz样本
dsa=$HOME/miniconda3/envs/download/etc/asperaweb_id_dsa.openssh
ls -lh $dsa
# conda activate download
# 自己搭建好 download 这个 conda 的小环境哦。
x=_1
y=_2
for id in {73..80}
do
ascp -QT -l 300m -P33001 -i $dsa \
era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/SRR123/0$id/SRR123031$id/SRR123031$id$x.fastq.gz .
ascp -QT -l 300m -P33001 -i $dsa \
era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/SRR123/0$id/SRR123031$id/SRR123031$id$y.fastq.gz .
done
把上面的代码存为代码文件: download.sh ,然后使用下面的命令放在后台下载即可:
conda activate download
nohup bash download.sh &
得到的文件如下:
1.2G 10月 9 22:07 SRR12303173_1.fastq.gz
1.2G 10月 9 22:12 SRR12303173_2.fastq.gz
1.1G 10月 9 22:17 SRR12303174_1.fastq.gz
1.2G 10月 9 22:24 SRR12303175_1.fastq.gz
1.3G 10月 9 22:30 SRR12303175_2.fastq.gz
1.1G 10月 10 09:54 SRR12303176_1.fastq.gz
1.1G 10月 10 09:56 SRR12303176_2.fastq.gz
1.4G 10月 10 10:02 SRR12303177_1.fastq.gz
1.4G 10月 10 10:05 SRR12303177_2.fastq.gz
1.2G 10月 10 10:09 SRR12303178_1.fastq.gz
1.2G 10月 10 10:13 SRR12303178_2.fastq.gz
1.2G 10月 10 10:18 SRR12303179_1.fastq.gz
1.2G 10月 10 10:21 SRR12303179_2.fastq.gz
1.3G 10月 10 10:26 SRR12303180_1.fastq.gz
1.3G 10月 10 10:35 SRR12303180_2.fastq.gz
可以看到,aspera下载的时候,中间11个小时任务终止了,是我自己重新跑了aspera下载,续起来了的。而且如果你仔细看会发现 SRR12303174这个样品只有R1的fq文件缺失了R2,也是需要重新单独下载。
conda activate download
dsa=$HOME/miniconda3/envs/download/etc/asperaweb_id_dsa.openssh
id=74;y=_2
ascp -QT -l 300m -P33001 -i $dsa \
era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/SRR123/0$id/SRR123031$id/SRR123031$id$y.fastq.gz .
## SRR12303174_2.fastq.gz 100% 1094MB 87.3Mb/s 01:27
## Completed: 1120670K bytes transferred in 87 seconds
## (104410K bits/sec), in 1 file.
## 全部文件下载完毕后,使用下面的命令检查一下fq.gz文件是否完整
gzip -t *gz
只有项目的fq数据全部准备而且确认无误后才能进行下一步!
测序数据的质量控制
这里选择trim_galore软件,自动批量运行:
mkdir -p ~/project/atac/
cd ~/project/atac/
conda activate atac
trim_galore --help
for id in {73..80}
do
nohup trim_galore -q 25 --phred33 --length 35 -e 0.1 --stringency 4 --paired \
-o clean raw/SRR123031$id*.fastq.gz &
done
得到的文件如下:
828M 10月 10 17:00 clean/SRR12303173_1_val_1.fq.gz
836M 10月 10 17:00 clean/SRR12303173_2_val_2.fq.gz
797M 10月 10 18:09 clean/SRR12303174_1_val_1.fq.gz
808M 10月 10 18:09 clean/SRR12303174_2_val_2.fq.gz
900M 10月 10 17:03 clean/SRR12303175_1_val_1.fq.gz
917M 10月 10 17:03 clean/SRR12303175_2_val_2.fq.gz
787M 10月 10 16:58 clean/SRR12303176_1_val_1.fq.gz
794M 10月 10 16:58 clean/SRR12303176_2_val_2.fq.gz
992M 10月 10 17:13 clean/SRR12303177_1_val_1.fq.gz
1006M 10月 10 17:13 clean/SRR12303177_2_val_2.fq.gz
845M 10月 10 17:01 clean/SRR12303178_1_val_1.fq.gz
855M 10月 10 17:01 clean/SRR12303178_2_val_2.fq.gz
840M 10月 10 17:01 clean/SRR12303179_1_val_1.fq.gz
847M 10月 10 17:01 clean/SRR12303179_2_val_2.fq.gz
945M 10月 10 17:06 clean/SRR12303180_1_val_1.fq.gz
963M 10月 10 17:06 clean/SRR12303180_2_val_2.fq.gz
这个过滤还是有点狠的,之前1.3G现在都小于1G了。实际上可以走fastqc+multiqc的质控看过滤前后的具体情况。
数据比对到参考基因组
1、mm10小鼠参考基因组的下载
#下载
mkdir -p ~/project/atac/ref
cd ~/project/atac/ref
nohup wget http://hgdownload.soe.ucsc.edu/goldenPath/mm10/bigZips/mm10.fa.gz &
#解压
gunzip mm10.fa.gz
2、bowtie2-build构建参考基因组索引文件
这一步会生成6个索引文件,这一步耗时比较常。也可以自行下载对应的参考基因组索引。
conda activate atac
nohup bowtie2-build mm10.fa mm10 1>log 2>&1 &
得到的文件如下:
848M 10月 10 17:20 mm10.1.bt2
633M 10月 10 17:20 mm10.2.bt2
6.0K 10月 10 16:36 mm10.3.bt2
633M 10月 10 16:36 mm10.4.bt2
2.6G 1月 23 2020 mm10.fa
848M 10月 10 18:05 mm10.rev.1.bt2
633M 10月 10 18:05 mm10.rev.2.bt2
3、bowtie2进行批量比对
首先制作配置文件:
cd ~/project/atac/align
ls $HOME/project/atac/clean/*_1.fq.gz > 1
ls $HOME/project/atac/clean/*_2.fq.gz > 2
ls $HOME/project/atac/clean/*_2.fq.gz |cut -d"/" -f 7|cut -d"_" -f 1 > 0
paste 0 1 2 > config.clean ## 供mapping使用的配置文件
然后创建含有如下内容的脚本(aligh.sh):
## 相对目录需要理解
bowtie2_index=$HOME/project/atac/ref/mm10
## 一定要搞清楚自己的bowtie2软件安装在哪里,以及自己的索引文件在什么地方!!!
#source activate atac
cat config.clean |while read id;
do echo $id
arr=($id)
fq2=${arr[2]}
fq1=${arr[1]}
sample=${arr[0]}
## 比对过程15分钟一个样本
bowtie2 -p 5 --very-sensitive -X 2000 -x $bowtie2_index -1 $fq1 -2 $fq2 |samtools sort -O bam -@ 5 -o - > ${sample}.raw.bam
samtools index ${sample}.raw.bam
bedtools bamtobed -i ${sample}.raw.bam > ${sample}.raw.bed
samtools flagstat ${sample}.raw.bam > ${sample}.raw.stat
# https://github.com/biod/sambamba/issues/177
sambamba markdup --overflow-list-size 600000 --tmpdir='./' -r ${sample}.raw.bam ${sample}.rmdup.bam
samtools index ${sample}.rmdup.bam
## ref:https://www.biostars.org/p/170294/
## Calculate %mtDNA:
mtReads=$(samtools idxstats ${sample}.rmdup.bam | grep 'chrM' | cut -f 3)
totalReads=$(samtools idxstats ${sample}.rmdup.bam | awk '{SUM += $3} END {print SUM}')
echo '==> mtDNA Content:' $(bc <<< "scale=2;100*$mtReads/$totalReads")'%'
samtools flagstat ${sample}.rmdup.bam > ${sample}.rmdup.stat
samtools view -h -f 2 -q 30 ${sample}.rmdup.bam |grep -v chrM |samtools sort -O bam -@ 5 -o - > ${sample}.last.bam
samtools index ${sample}.last.bam
samtools flagstat ${sample}.last.bam > ${sample}.last.stat
bedtools bamtobed -i ${sample}.last.bam > ${sample}.bed
done
提交脚本的代码是:
conda activate atac
nohup bash aligh.sh 1>log 2>&1 &
全部运行完毕后输出非常多文件。
首先看bam文件,如下:
1.1G 10月 11 15:49 SRR12303173.last.bam
1.8G 10月 10 23:01 SRR12303173.raw.bam
1.3G 10月 11 15:48 SRR12303173.rmdup.bam
823M 10月 11 16:00 SRR12303174.last.bam
1.7G 10月 11 00:24 SRR12303174.raw.bam
976M 10月 11 15:59 SRR12303174.rmdup.bam
1.4G 10月 11 16:11 SRR12303175.last.bam
2.2G 10月 11 02:26 SRR12303175.raw.bam
1.6G 10月 11 16:09 SRR12303175.rmdup.bam
1.2G 10月 11 16:23 SRR12303176.last.bam
1.8G 10月 11 03:52 SRR12303176.raw.bam
1.4G 10月 11 16:21 SRR12303176.rmdup.bam
1.8G 10月 11 16:37 SRR12303177.last.bam
2.5G 10月 11 06:16 SRR12303177.raw.bam
2.1G 10月 11 16:35 SRR12303177.rmdup.bam
1.2G 10月 11 16:50 SRR12303178.last.bam
1.9G 10月 11 07:53 SRR12303178.raw.bam
1.4G 10月 11 16:48 SRR12303178.rmdup.bam
1.2G 10月 11 17:02 SRR12303179.last.bam
1.9G 10月 11 09:35 SRR12303179.raw.bam
1.4G 10月 11 17:00 SRR12303179.rmdup.bam
1.7G 10月 11 17:16 SRR12303180.last.bam
2.4G 10月 11 11:51 SRR12303180.raw.bam
1.9G 10月 11 17:14 SRR12303180.rmdup.bam
每个样品分别会输出3个bam文件,测序数据比对的bam,以及去除PCR重复后的bam,以及去除线粒体reads后的bam文件。
查看log日志,发现这些样本的线粒体含量是:
==> mtDNA Content: 1.81%
==> mtDNA Content: 3.72%
==> mtDNA Content: 1.88%
==> mtDNA Content: 1.98%
==> mtDNA Content: 2.16%
==> mtDNA Content: 3.78%
==> mtDNA Content: 2.11%
==> mtDNA Content: 2.17%
因为我们是首先去除PCR重复然后计算线粒体含量,其实是不准确的。
比对后的bam文件的统计
测序文库复杂度的检验
一个简单的含有awk脚本的shell脚本,代码如下:
ls *.last.bam|while read id;
do
bedtools bamtobed -bedpe -i $id | \
awk 'BEGIN{OFS="\t"}{print $1,$2,$4,$6,$9,$10}' | sort | uniq -c | \
awk 'BEGIN{mt=0;m0=0;m1=0;m2=0} ($1==1){m1=m1+1} ($1==2){m2=m2+1} {m0=m0+1} {mt=mt+$1} END{m1_m2=-1.0; if(m2>0) m1_m2=m1/m2;printf "%d\t%d\t%d\t%d\t%f\t%f\t%f\n",mt,m0,m1,m2,m0/mt,m1/m0,m1_m2}' > ${id%%.*}.nodup.pbc.qc;
done
脚本制作好了后命名为:
conda activate atac
nohup bash stat_qc.sh &
Library complexity measures计算结果如下,…nodup.pbc.qc文件格式为:
TotalReadPairs
DistinctReadPairs
OneReadPair
TwoReadPairs
NRF=Distinct/Total
PBC1=OnePair/Distinct
PBC2=OnePair/TwoPair
针对NRF、PBC1、PBC2这几个指标,ENCODE官网提供了标准.
计算结果显示NRF、PBC1、PBC2的值都非常完美,说明我们进行过滤和PCR去重的bam文件质量上没有问题,可以用于后续的分析。
前面的步骤是为了输出 last.bam 的文件,需要首先转为tagAlign,然后作为macs的输入文件去找peaks,拿到peaks后进行注释。
另外,后面的步骤我们换了一个课题,但是分析内容是一致的,我把aspera下载的代码同样的共享在这里哈:
x=_1
y=_2
for id in {93,98,99}
do
ascp -QT -l 300m -P33001 -i $dir era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/SRR126/0$id/SRR126920$id/SRR126920$id$x.fastq.gz .
ascp -QT -l 300m -P33001 -i $dir era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/SRR126/0$id/SRR126920$id/SRR126920$id$y.fastq.gz .
done
五、生成tagAlign格式文件
1. Convert PE BAM to tagAlign
- 对于单端序列。直接用bed格式就可以;对于双端序列,推荐用bedpe格式。这两种格式都可以称之为tagAlign,可以作为macs的输入文件。
- tagAligen格式相比bam,文件大小会小很多,更加方便文件的读取。在转换得到tagAlign格式之后,我们就可以很容易的将坐标进行偏移
nohup ls *nodup.srt.name.bam|while read id; do bedtools bamtobed -bedpe -mate1 -i $id | gzip -nc > ${id%%.*}.nodup.srt.name.bedpe.gz;done &
#含有chrM的染色体的TagAlign文件
nohup ls *.nodup.srt.name.bedpe.gz | while read id; do zcat $id | awk 'BEGIN{OFS="\t"}{printf "%s\t%s\t%s\tN\t1000\t%s\n%s\t%s\t%s\tN\t1000\t%s\n",$1,$2,$3,$9,$4,$5,$6,$10}' | gzip -nc > ${id%%.*}.nodup.srt.name.tagAlign.gz; done &
#去除chrM的染色体的TagAlign文件
nohup ls *nodup.srt.name.bedpe.gz|while read id; do zcat $id | grep -P -v "^chrM" | awk 'BEGIN{OFS="\t"}{printf "%s\t%s\t%s\tN\t1000\t%s\n%s\t%s\t%s\tN\t1000\t%s\n",$1,$2,$3,$9,$4,$5,$6,$10}' | gzip -nc > ${id%%.*}.nodup.nomit.srt.name.tagAlign.gz; done
2. Stand Cross Correlation analysis
| 用于评估ATAC/Chip实验质量好坏的一个重要指标 |
NREADS=25000000
nohup ls *.nodup.srt.name.bedpe.gz | while read id; do zcat $id | grep -v “chrM” | shuf -n ${NREADS} --random-source=<(openssl enc -aes-256-ctr -pass pass:$(zcat -f ${id%%.*}.nodup.srt.name.tagAlign.gz | wc -c) -nosalt </dev/zero 2>/dev/null) | awk 'BEGIN{OFS="\t"}{print $1,$2,$3,"N","1000",$9}' | gzip -nc > ${id%%.*}.nodup.nomit.srt.name.$((NREADS / 1000000)).tagAlign.gz; done &
#命令最终会生成交叉相关质量评估文件,*.cc.qc文件中会输出包含11列的信息,重点关注9-11列的信息,cc.plot.pdf文件相当于*.cc.qc文件的可视化
nohup ls *$((NREADS / 1000000)).tagAlign.gz | while read id; do Rscript $(which run_spp.R) -c=$id -p=10 -filtchr=chrM -savp=${id%%.*}.cc.plot.pdf -out=${id%%.*}.cc.qc; done &
#质控结果查看,主要看NSC,RSC,Quality tag三个值即输出文件的第9列,第10列,第11列。
ls *.cc.qc|while read id; do cat $id | awk '{print $9, "\t", $10, "\t", $11}';done
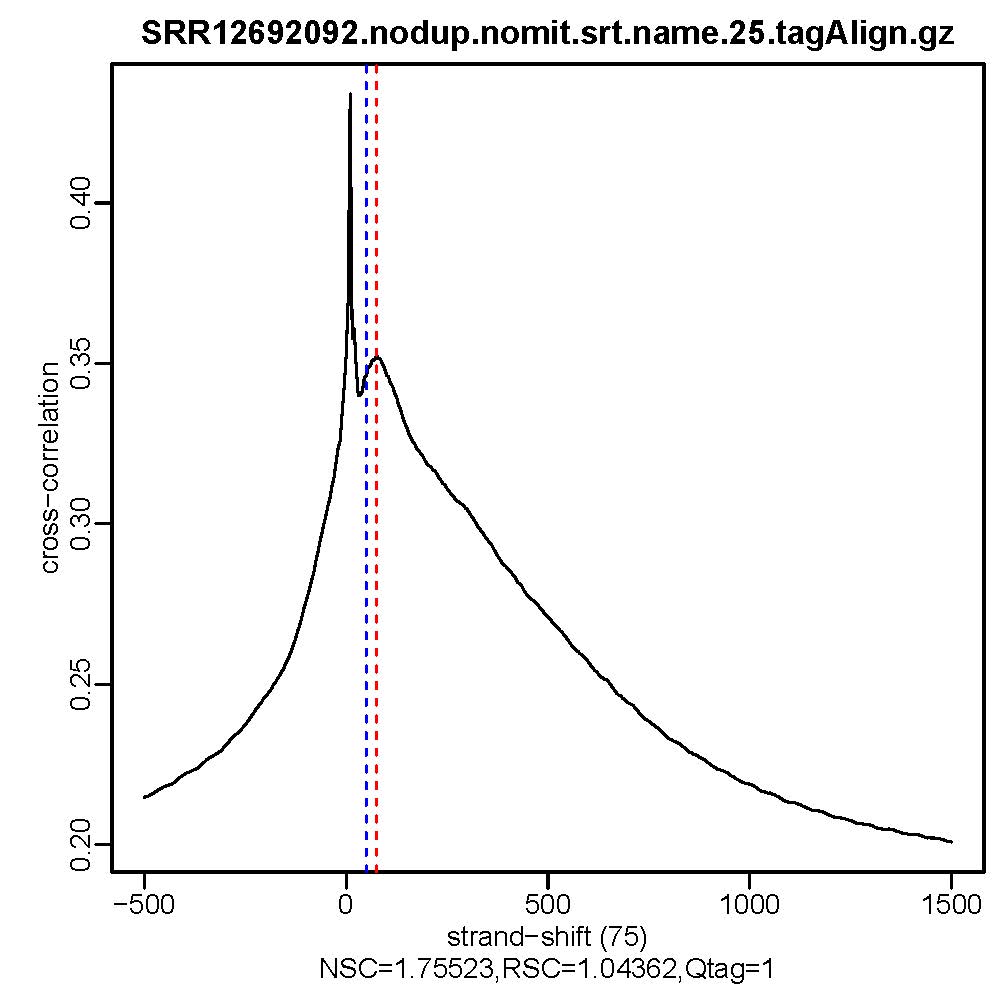
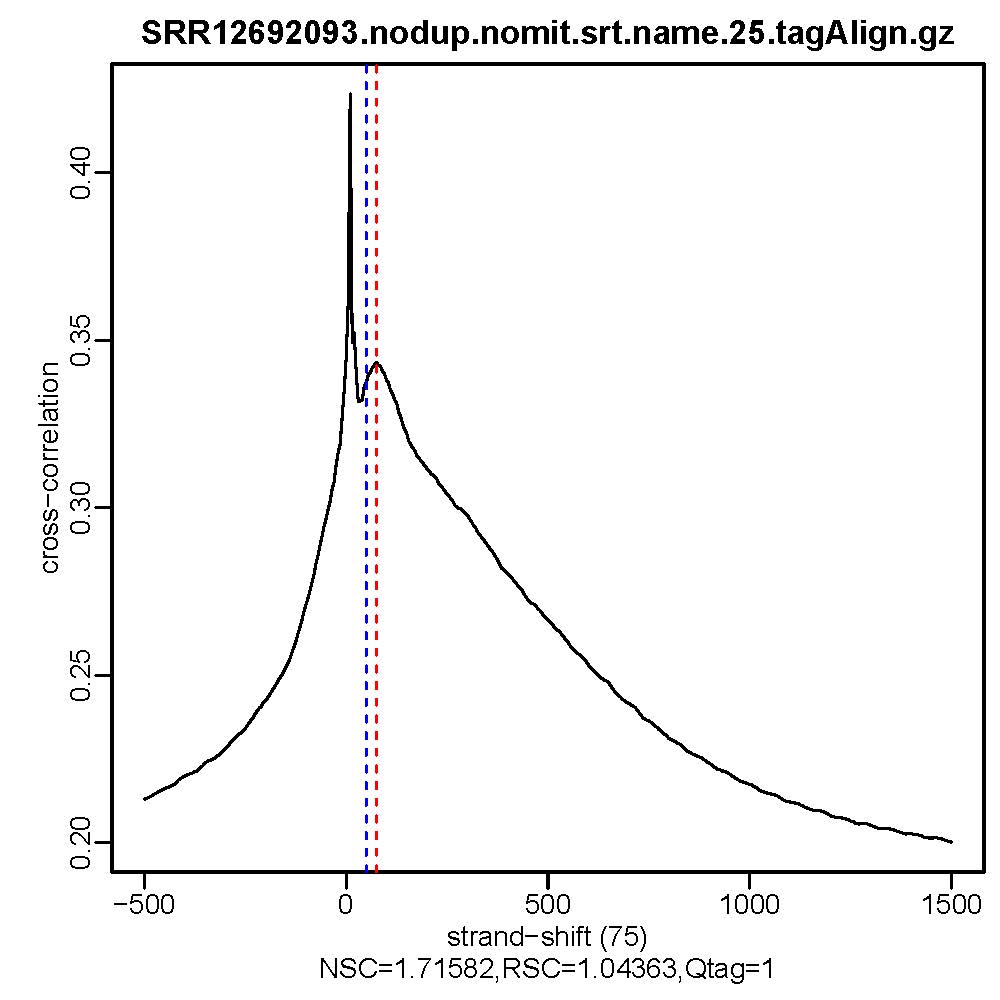
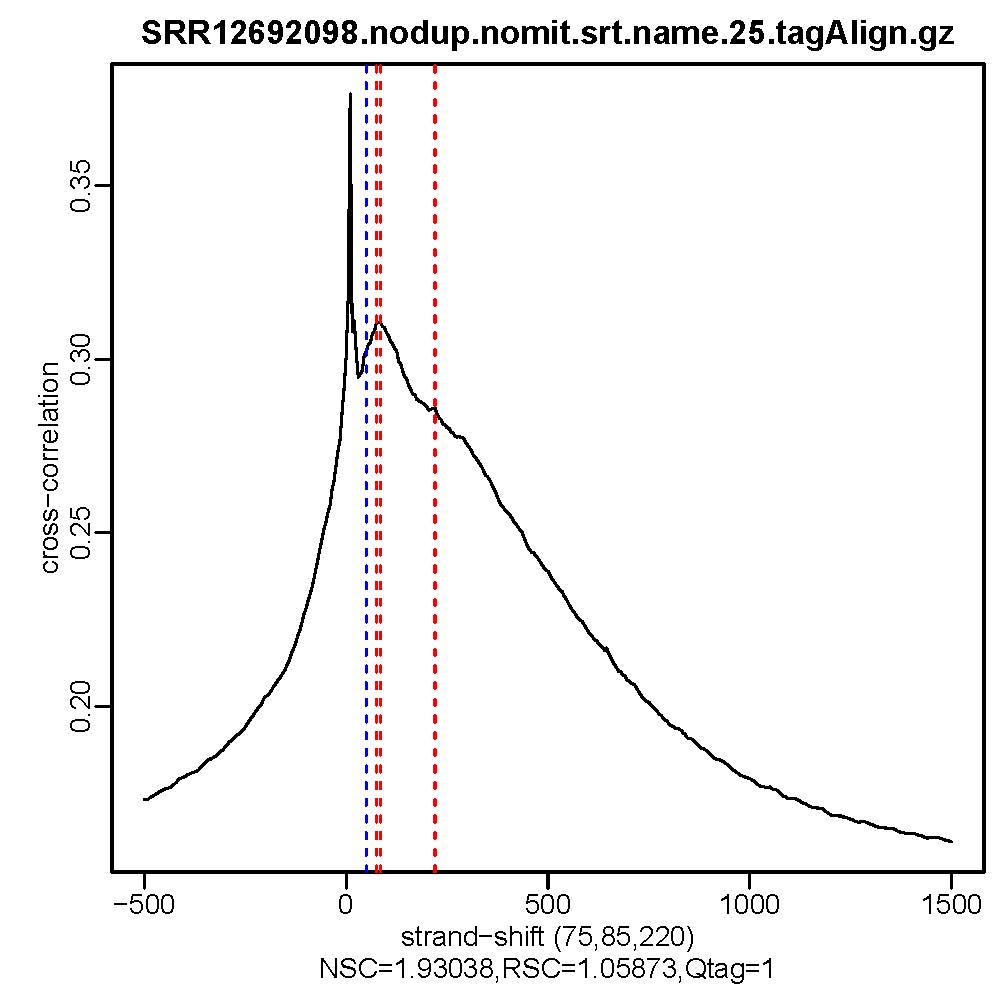

- 质控结果解读
Normalized strand cross-correlation coefficent (NSC):NSC是最大交叉相关值除以背景交叉相关的比率(所有可能的链转移的最小交叉相关值)。NSC值越大表明富集效果越好,NSC值低于1.1表明较弱的富集,小于1表示无富集。NSC值稍微低于1.05,有较低的信噪比或很少的峰,这肯能是生物学真实现象,比如有的因子在特定组织类型中只有很少的结合位点;也可能确实是数据质量差。
Relative strand cross-correlation coefficient (RSC):RSC是片段长度相关值减去背景相关值除以phantom-peak相关值减去背景相关值。RSC的最小值可能是0,表示无信号;富集好的实验RSC值大于1;低于1表示质量低。
QualityTag: Quality tag based on thresholded RSC (codes: -2:veryLow,-1:Low,0:Medium,1:High,2:veryHigh)
查看交叉相关性质量评估结果,主要看NSC,RSC,Quality tag三个值,这三个值分别对应输出文件的第9列,第10列,第11列。
六、Call Peaks
1、去除线粒体基因的TagAlign格式文件进行shift操作,输入macs2软件去callpeak
smooth_window=150 # default
shiftsize=$(( -$smooth_window/2 ))
pval_thresh=0.01
nohup ls *nodup.nomit.srt.name.tagAlign.gz | while read id; \
do macs2 callpeak \
-t $id -f BED -n "${id%%.*}" -g mm -p $pval_thresh \
--shift $shiftsize --extsize $smooth_window --nomodel -B --SPMR --keep-dup all --call-summits; \
done &
2、去除ENCODE列入黑名单的区域
- 去除黑名单的bed文件,用于后续的peaks注释
BLACKLIST=/home/gongyuqi/project/ATAC/mm10.blacklist.bed.gz
#*_summits.bed为macs2软件callpeak的结果文件之一
nohup ls *_summits.bed | while read id; do bedtools intersect -a $id -b $BLACKLIST -v > ${id%%.*}_filt_blacklist.bed; done &
#查看过滤黑名单的区域前后的bed文件的peaks数
ls *summits.bed|while read id; do cat $id |wc -l >>summits.bed.txt;done
ls *summits_filt_blacklist.bed|while read id; do cat $id |wc -l >>summits_filt_blacklist.bed.txt;done
past summits.bed.txt summits_filt_blacklist.bed.txt
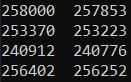
- 去除黑名单的narrowPeaks文件,用于后续的IDR评估
#使用IDR需要先对MACS2的结果文件narrowPeak根据-log10(p-value)进行排序,-log10(p-value)在第八列。
# Sort by Col8 in descending order and replace long peak names in Column 4 with Peak_<peakRank>
#*_peaks.narrowPeak为macs2软件callpeak的结果文件之一
NPEAKS=300000
ls *_peaks.narrowPeak | while read id; do sort -k 8gr,8gr $id | awk 'BEGIN{OFS="\t"}{$4="Peak_"NR ; print $0}' | head -n ${NPEAKS} | gzip -nc > ${id%%_*}.narrowPeak.gz; done
BLACKLIST=../BLACKLIST/mm10.blacklist.bed.gz
#生成不压缩文件
ls *.narrowPeak.gz | while read id; do bedtools intersect -v -a $id -b ${BLACKLIST} | awk 'BEGIN{OFS="\t"} {if ($5>1000) $5=1000; print $0}' | grep -P 'chr[\dXY]+[ \t]' > ${id%%.*}.narrowPeak.filt_blacklist; done
#生成压缩文件
#ls *.narrowPeak.gz | while read id; do bedtools intersect -v -a $id -b ${BLACKLIST} | awk 'BEGIN{OFS="\t"} {if ($5>1000) $5=1000; print $0}' | grep -P 'chr[\dXY]+[ \t]' | gzip -nc > ${id%%.*}.narrowPeak.filt_blacklist.gz; done
3、Irreproducibility Discovery Rate (IDR)评估
| 用于评估重复样本间peaks一致性的重要指标 |
首先生成narrowPeak_sample.txt文件,方便后续循环处理,生成文件内容如下:

nohup cat narrowPeak_sample.txt | while read id
do
arr=(${id})
Rep1=${arr[0]}
Rep2=${arr[1]}
sample=${Rep1%%.*}_${Rep2%%.*}_idr
idr --samples $Rep1 $Rep2 --input-file-type narrowPeak -o $sample --plot
done &
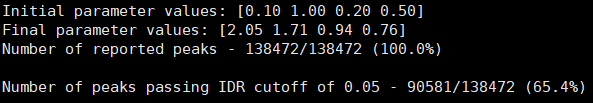
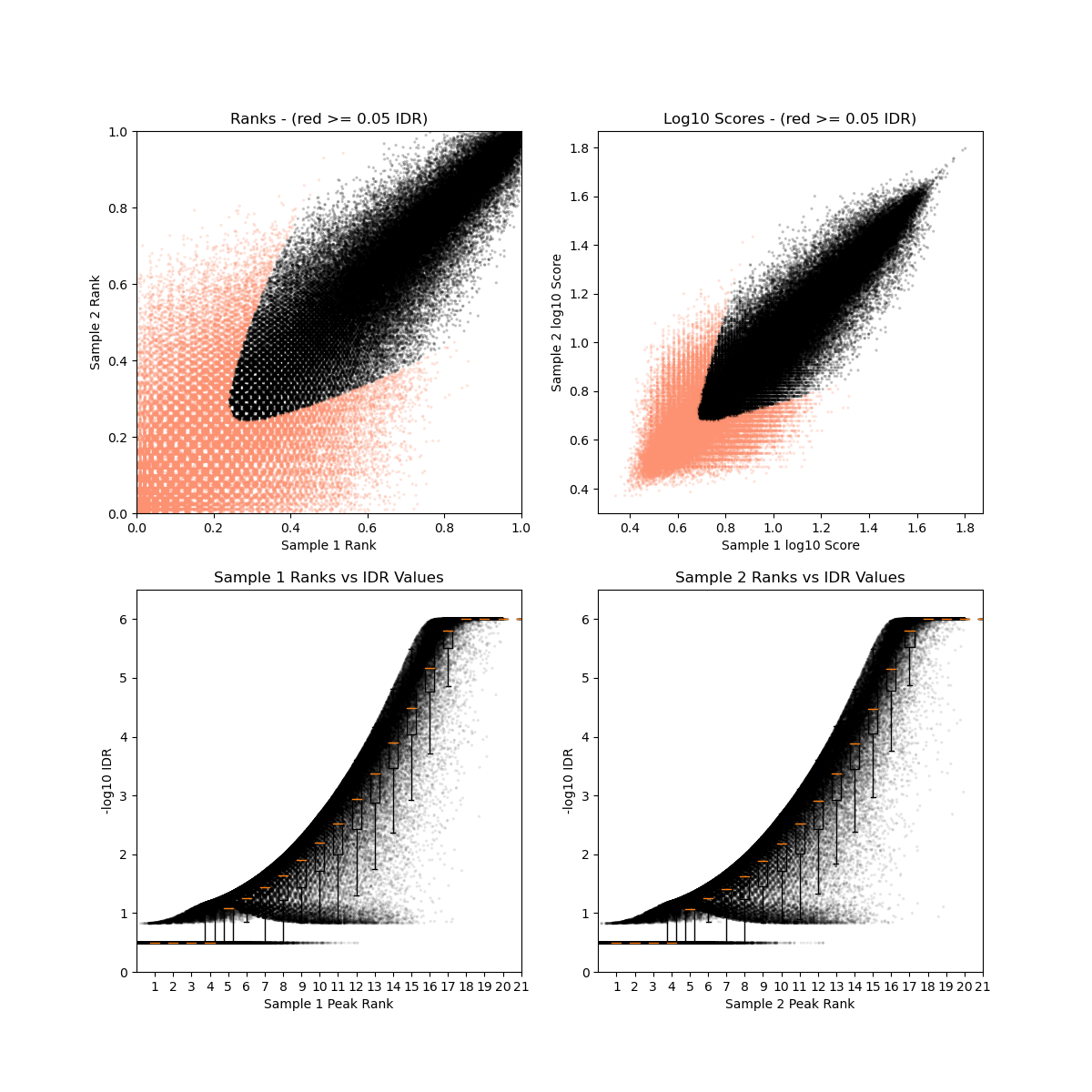
- DMSO_24h_wt (样本处理情况)
- SRR12692092.filt_blacklist.narrowPeak SRR12692093.filt_blacklist.narrowPeak
- 没有通过IDR阈值的显示为红色
- BRM014-10uM_24h_wt (样本处理情况)
- SRR12692098.filt_blacklist.narrowPeak SRR12692099.filt_blacklist.narrowPeak
- 没有通过IDR阈值的显示为红色
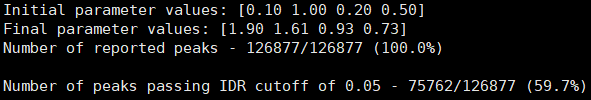

IDR评估会同时考虑peaks间的overlap和富集倍数的一致性。通过IDR阈值(0.05)的占比越大,说明重复样本间peaks一致性越好。从idr的分析结果看,我们的测试数据还可以的呢。
IDR评估相关参考资料:
4、Fraction of reads in peaks (FRiP)评估
| 反映样本富集效果好坏的评价指标 |
#生成bed文件
nohup ls *.nodup.bam|while read id;do (bedtools bamtobed -i $id >${id%%.*}.nodup.bed) ;done &
#批量计算FRiP
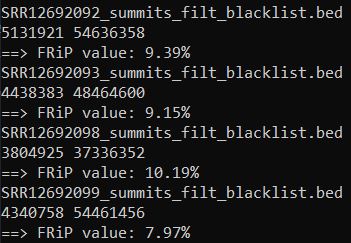
ls *_summits_filt_blacklist.bed|while read id;
do
echo $id
bed=${id%%_*}.nodup.bed
Reads=$(bedtools intersect -a $bed -b $id |wc -l|awk '{print $1}')
totalReads=$(wc -l $bed|awk '{print $1}')
echo $Reads $totalReads
echo '==> FRiP value:' $(bc <<< "scale=2;100*$Reads/$totalReads")'%'
done
FRiP值在5%以上算比较好的。但也不绝对,这是个软阈值,可以作为一个参考。
FRiP评估相关参考资料:
七、Peak annotation
1、Feature Distribution
setwd("path to bed file")
library(ChIPpeakAnno)
library(TxDb.Mmusculus.UCSC.mm10.knownGene)
library(org.Mm.eg.db)
library(BiocInstaller)
library(ChIPseeker)
txdb <- TxDb.Mmusculus.UCSC.mm10.knownGene
promoter <- getPromoters(TxDb=txdb, upstream=3000, downstream=3000)
files = list(DMSO_24h_wt_rep1 = "SRR12692092_summits_filt_blacklist.bed",
DMSO_24h_wt_rep2 = "SRR12692093_summits_filt_blacklist.bed",
BRM014_10uM_24h_wt_rep1 = "SRR12692098_summits_filt_blacklist.bed",
BRM014_10uM_24h_wt_rep2 = "SRR12692099_summits_filt_blacklist.bed")
#汇总所有样本
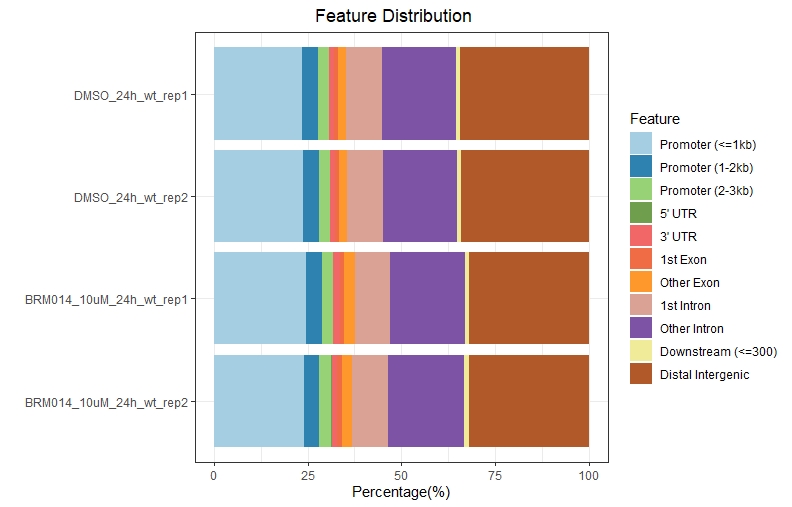
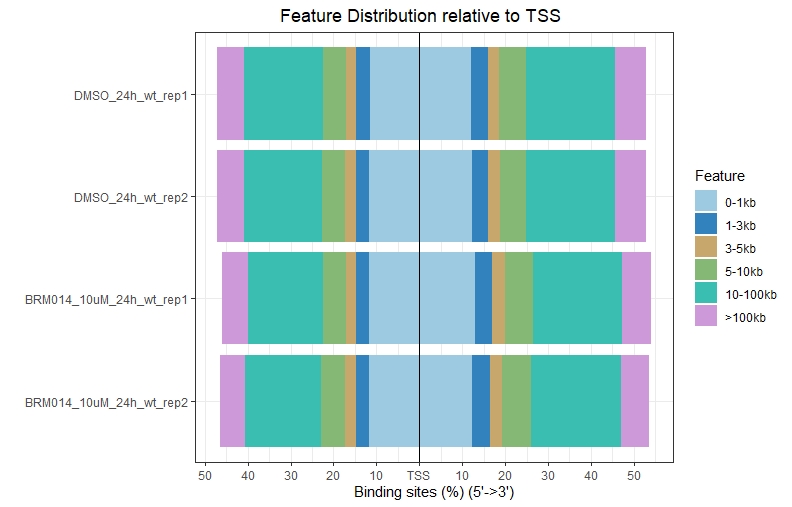
#plotAnnoBar和plotDistToTSS这两个柱状图都支持多个数据同时展示
peakAnnoList <- lapply(files, annotatePeak,
TxDb=txdb,
tssRegion=c(-3000, 3000))
plotAnnoBar(peakAnnoList,title = " Feature Distribution")
plotDistToTSS(peakAnnoList,title = " Feature Distribution relative to TSS")
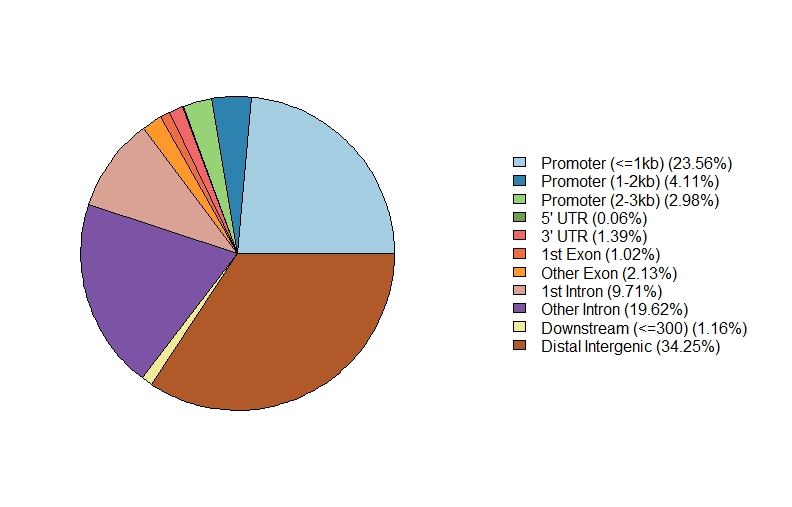
#例举单个样本
peakAnno <- annotatePeak(files[[1]],# 分别改成2或者3或者4即可,分别对应四个文件
tssRegion=c(-3000, 3000),
TxDb=txdb,
annoDb="org.Mm.eg.db")
plotAnnoPie(peakAnnoLipeakAnnost)
upsetplot(peakAnno, vennpie=TRUE)
.jpeg)
2、查看peaks在全基因组上的分布
#输入文件的准备
DMSO_24h_wt_rep1<-read.csv("SRR12692092_summits_filt_blacklist.csv")
DMSO_24h_wt_rep1<-DMSO_24h_wt_rep1[,-4]
DMSO_24h_wt_rep2<-read.csv("SRR12692093_summits_filt_blacklist.csv")
DMSO_24h_wt_rep2<-DMSO_24h_wt_rep2[,-4]
BRM014_10uM_24h_wt_rep1<-read.csv("SRR12692098_summits_filt_blacklist.csv")
BRM014_10uM_24h_wt_rep1<-BRM014_10uM_24h_wt_rep1[,-4]
BRM014_10uM_24h_wt_rep2<-read.csv("SRR12692099_summits_filt_blacklist.csv")
BRM014_10uM_24h_wt_rep2<-BRM014_10uM_24h_wt_rep2[,-4]
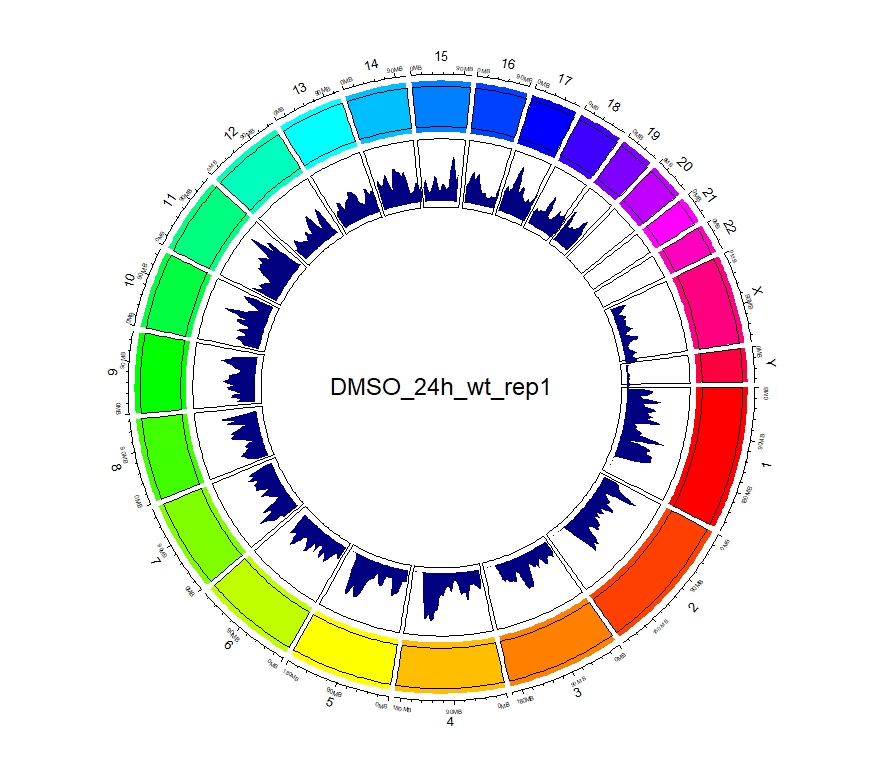
#以DMSO_24h_wt_rep1为例
set.seed(123)
circos.initializeWithIdeogram(plotType = c("axis", "labels"))
circos.track(ylim = c(0, 1), panel.fun = function(x, y) {
chr = CELL_META$sector.index
xlim = CELL_META$xlim
ylim = CELL_META$ylim
circos.rect(xlim[1], 0, xlim[2], 1)
}, track.height = 0.15, bg.border = NA, bg.col=rainbow(24))
text(0, 0, "DMSO_24h_wt_rep1", cex = 1.5)
circos.genomicDensity(DMSO_24h_wt_rep1, col = c("#000080"), track.height = 0.2)
circos.clear()
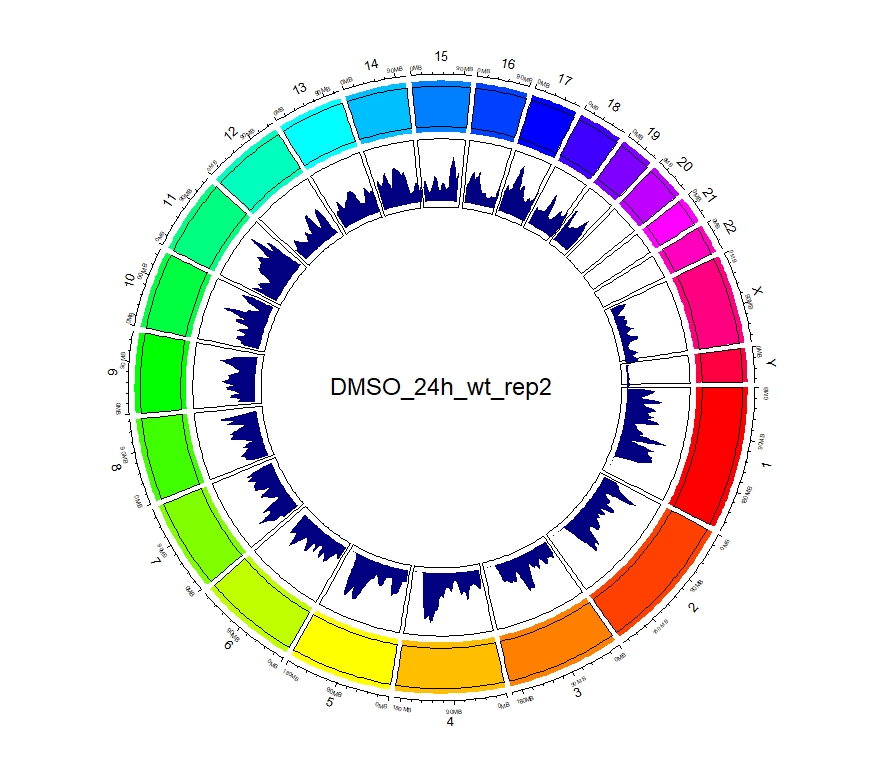
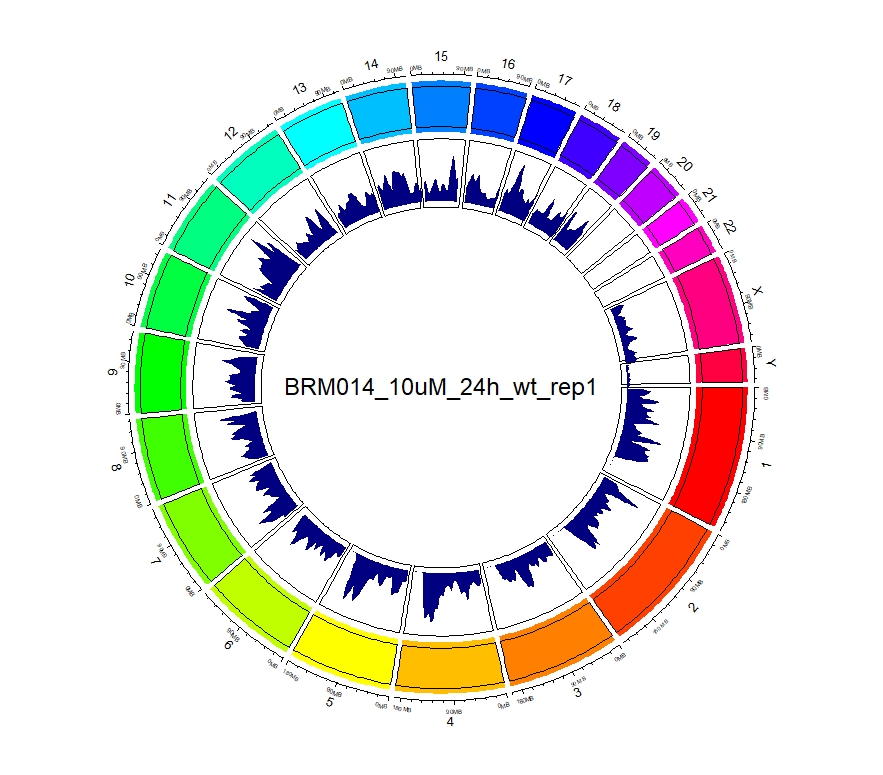
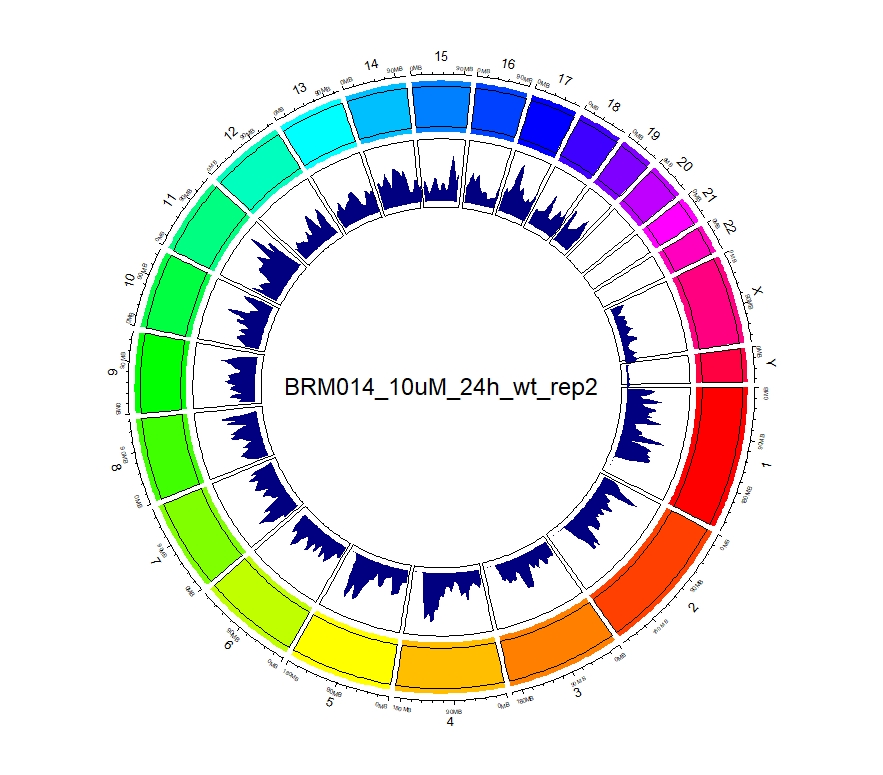
看到这样的结果,第一反应就是————为什么两种处理情况下染色体开放程度那么像!?难道我代码有问题!?经过反复检查验证(将一个样本chr1上的peaks都删掉,再次运行上述代码,就会发现显著的改变),可以确定分析上是没有问题的。这两种处理导致的差异可能不是很显著。再加上20万+的peaks放在这个小小的circos图上展示,有些差异会被掩盖掉。就如在做TSS富集分析的时候,单独看TSS前后3Kb区域,可以看到有两个峰,但在看TSS-genebody-TSE区域是,TSS处相对微弱的那个峰就被掩盖掉了。
3、拿到每个样本中peaks对应得基因名
这一步非常重要,拿到基因名就可以根据课题需要进行差异分析等
#以DMSO_24h_wt样本为例
#replicate 1
peakAnno_DMSO_24h_wt_rep1 <- annotatePeak(files[[1]],
tssRegion=c(-3000, 3000),
TxDb=txdb,
annoDb="org.Mm.eg.db")
genelist_DMSO_24h_wt_rep1_uniqe<-as.data.frame(unique(peakAnno_DMSO_24h_wt_rep1@anno@elementMetadata@listData[["SYMBOL"]]))
colnames(genelist_DMSO_24h_wt_rep1_uniqe)<-"symbol"
#replicate 2
peakAnno_DMSO_24h_wt_rep2 <- annotatePeak(files[[2]],
tssRegion=c(-3000, 3000),
TxDb=txdb,
annoDb="org.Mm.eg.db")
genelist_DMSO_24h_wt_rep2_uniqe<-as.data.frame(unique(peakAnno_DMSO_24h_wt_rep2@anno@elementMetadata@listData[["SYMBOL"]]))
colnames(genelist_DMSO_24h_wt_rep2_uniqe)<-"symbol"
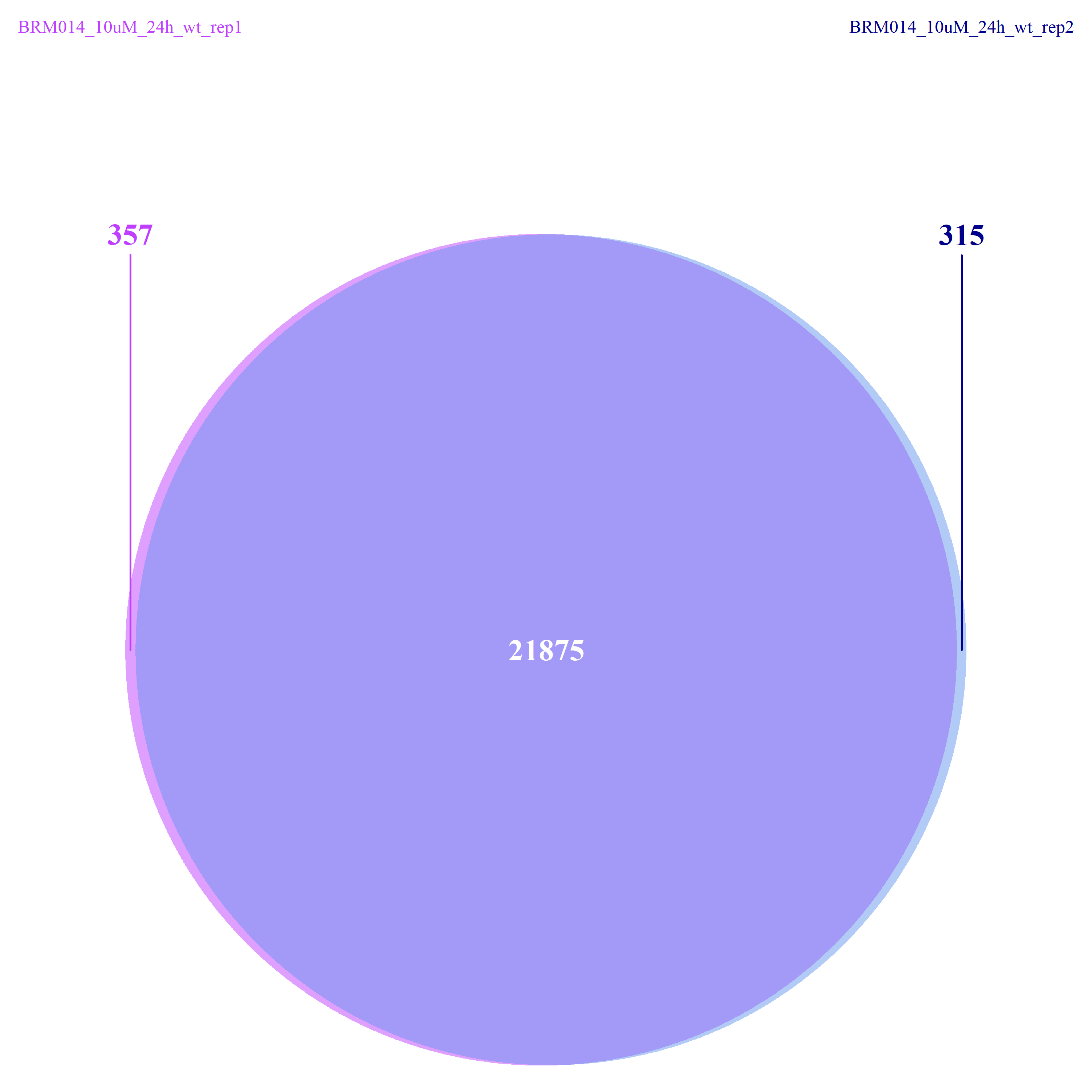
#重复样本间共同的开放基因
venn.diagram(
x=list(
DMSO_24h_wt_rep1=genelist_DMSO_24h_wt_rep1_uniqe$symbol,
DMSO_24h_wt_rep2=genelist_DMSO_24h_wt_rep2_uniqe$symbol
),
filename = "DMSO_24h_wt.png",
lty="dotted",
lwd=3,
col="transparent",
fill=c("darkorchid1","cornflowerblue"),
alpha=0.5,
label.col=c("darkorchid1","white","darkblue") ,
cex=1,
fontfamily="serif",
fontface="bold",
cat.default.pos="text",
cat.col=c("darkorchid1","darkblue"),
cat.cex=0.6,
cat.fontfamily="serif",
cat.dist=c(0.3,0.3),
cat.pos=0
)
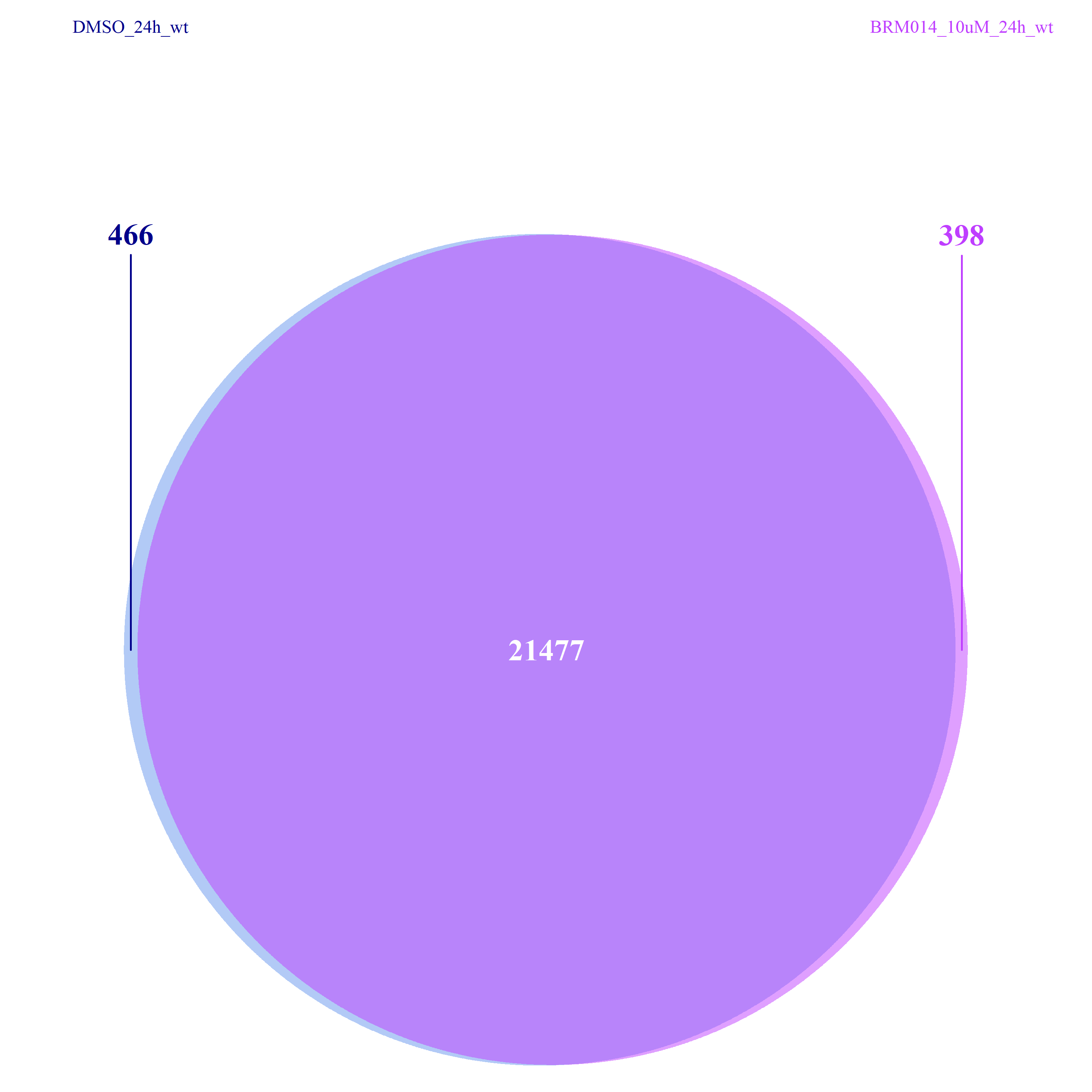
#查看各组内样本间的overlapping reads:DMSO_24h_wt, BRM014_10uM_24h_wt;
#以及组间peaks的异同情况:DMSO_24h_wt vs. BRM014_10uM_24h_wt
#代码类似上面的,就不一一展示了
从下图可以看出,不管是组间还是组内,差异的peaks数目都不是很多了,这一点也验证了我们上面做的再全基因组范围内查看peaks的分布结果。

网页工具绝对是完成不了这样的命令行数据分析哦
这个是基于Linux的ngs数据的上游处理,目前没有成熟的网页工具支持这样的分析。其实呢,如果你有时间请务必学习编程基础,自由自在的探索海量的公共数据,辅助你的科研,那么:
如果你没有时间从头开始学编程,也可以委托专业的团队付费拿到同样的数据分析, 比如我们。一条龙服务,一个简单的ATAC-seq项目的标准分析(从fq文件到peaks的注释)仅收费1600,而且是可以拿到全部的数据和代码哦!
如果TAC-seq项目实验设计比较复杂,比如多个实验条件多个时间点,需要做差异分析或者时序分析,费用会比较高昂,请谨慎联系我们哈!
- 需要自己读文献筛选合适的数据集
- 提供1个小时左右的一对一讲解转录组数据处理背景知识。
如果需要委托,直接在我们《生信技能树》公众号留言即可,我们会安排合适的生信工程师对接具体的项目。