有粉丝在我们公众号后台咨询,基因名字没有什么规律,自己拿到了一堆基因不知道如何是好,希望我们给一个案例。
他拿到了基因列表如下
简单的列出来给大家看看:
AR
ARID3A
ATF1
BHLHE40
CDK9
CEBPA
CEBPB
CEBPD
CHD8
CREB1
CREBBP
ELF1
EP300
ESR1
FOSL2
FOXA1
FOXA2
GABPA
HDAC2
HEY1
HNF4A
HNF4G
JUND
KDM5B
MAX
MAZ
MBD2
MBD4
MXI1
MYBL2
NFIC
NR2C2
NR2F2
POLR2A
RAD21
REST
RXRA
SIN3A
SP1
SPI1
SREBF1
TAF1
TBP
TCF12
TRIM24
TRIM28
USF1
YY1
ZBTB7A
ZEB1
这样的基因名字,很难让大家知道其背后的生物学意义!所以我们需要对它进行各式各样的处理。
把这些基因名字保存在 genelist.txt 文件里面,后面就交给R语言啦。
第一步:看基因名字
代码是:
rm(list = ls()) ## 魔幻操作,一键清空~
options(stringsAsFactors = F)
library(survminer)
library(GEOquery)
surGenes=read.table('genelist.txt')[,1]
head(surGenes)
library(ggplot2)
library(clusterProfiler)
library(org.Hs.eg.db)
df <- bitr(unique(surGenes), fromType = "SYMBOL",
toType = c( "ENTREZID" ,'GENENAME'),
OrgDb = org.Hs.eg.db)
head(df)
df
# https://biit.cs.ut.ee/gprofiler/convert
gene_up=df$ENTREZID
拿到基因名字等信息,如下:
SYMBOL ENTREZID GENENAME
1 AR 367 androgen receptor
2 ARID3A 1820 AT-rich interaction domain 3A
3 ATF1 466 activating transcription factor 1
4 BHLHE40 8553 basic helix-loop-helix family member e40
5 CDK9 1025 cyclin dependent kinase 9
6 CEBPA 1050 CCAAT enhancer binding protein alpha
7 CEBPB 1051 CCAAT enhancer binding protein beta
8 CEBPD 1052 CCAAT enhancer binding protein delta
9 CHD8 57680 chromodomain helicase DNA binding protein 8
10 CREB1 1385 cAMP responsive element binding protein 1
11 CREBBP 1387 CREB binding protein
12 ELF1 1997 E74 like ETS transcription factor 1
13 EP300 2033 E1A binding protein p300
14 ESR1 2099 estrogen receptor 1
15 FOSL2 2355 FOS like 2, AP-1 transcription factor subunit
16 FOXA1 3169 forkhead box A1
17 FOXA2 3170 forkhead box A2
18 GABPA 2551 GA binding protein transcription factor subunit alpha
19 HDAC2 3066 histone deacetylase 2
20 HEY1 23462 hes related family bHLH transcription factor with YRPW motif 1
21 HNF4A 3172 hepatocyte nuclear factor 4 alpha
22 HNF4G 3174 hepatocyte nuclear factor 4 gamma
23 JUND 3727 JunD proto-oncogene, AP-1 transcription factor subunit
24 KDM5B 10765 lysine demethylase 5B
25 MAX 4149 MYC associated factor X
26 MAZ 4150 MYC associated zinc finger protein
27 MBD2 8932 methyl-CpG binding domain protein 2
28 MBD4 8930 methyl-CpG binding domain 4, DNA glycosylase
29 MXI1 4601 MAX interactor 1, dimerization protein
30 MYBL2 4605 MYB proto-oncogene like 2
31 NFIC 4782 nuclear factor I C
32 NR2C2 7182 nuclear receptor subfamily 2 group C member 2
33 NR2F2 7026 nuclear receptor subfamily 2 group F member 2
34 POLR2A 5430 RNA polymerase II subunit A
35 RAD21 5885 RAD21 cohesin complex component
36 REST 5978 RE1 silencing transcription factor
37 RXRA 6256 retinoid X receptor alpha
38 SIN3A 25942 SIN3 transcription regulator family member A
39 SP1 6667 Sp1 transcription factor
40 SPI1 6688 Spi-1 proto-oncogene
41 SREBF1 6720 sterol regulatory element binding transcription factor 1
42 TAF1 6872 TATA-box binding protein associated factor 1
43 TBP 6908 TATA-box binding protein
44 TCF12 6938 transcription factor 12
45 TRIM24 8805 tripartite motif containing 24
46 TRIM28 10155 tripartite motif containing 28
47 USF1 7391 upstream transcription factor 1
48 YY1 7528 YY1 transcription factor
49 ZBTB7A 51341 zinc finger and BTB domain containing 7A
50 ZEB1 6935 zinc finger E-box binding homeobox 1
第二步:kegg数据库注释
代码是:
enrichKK <- enrichKEGG(gene = gene_up,
organism = 'hsa',
#universe = gene_all,
pvalueCutoff = 0.1,
qvalueCutoff =0.1)
head(enrichKK)[,1:6]
dotplot(enrichKK)
enrichKK=DOSE::setReadable(enrichKK, OrgDb='org.Hs.eg.db',keyType='ENTREZID')
enrichKK
tmp=enrichKK@result
里面的enrichKEGG函数会Reading KEGG annotation online:,在中国大陆部分地区会失败哦,或者网络很差。
如下所示注释结果:
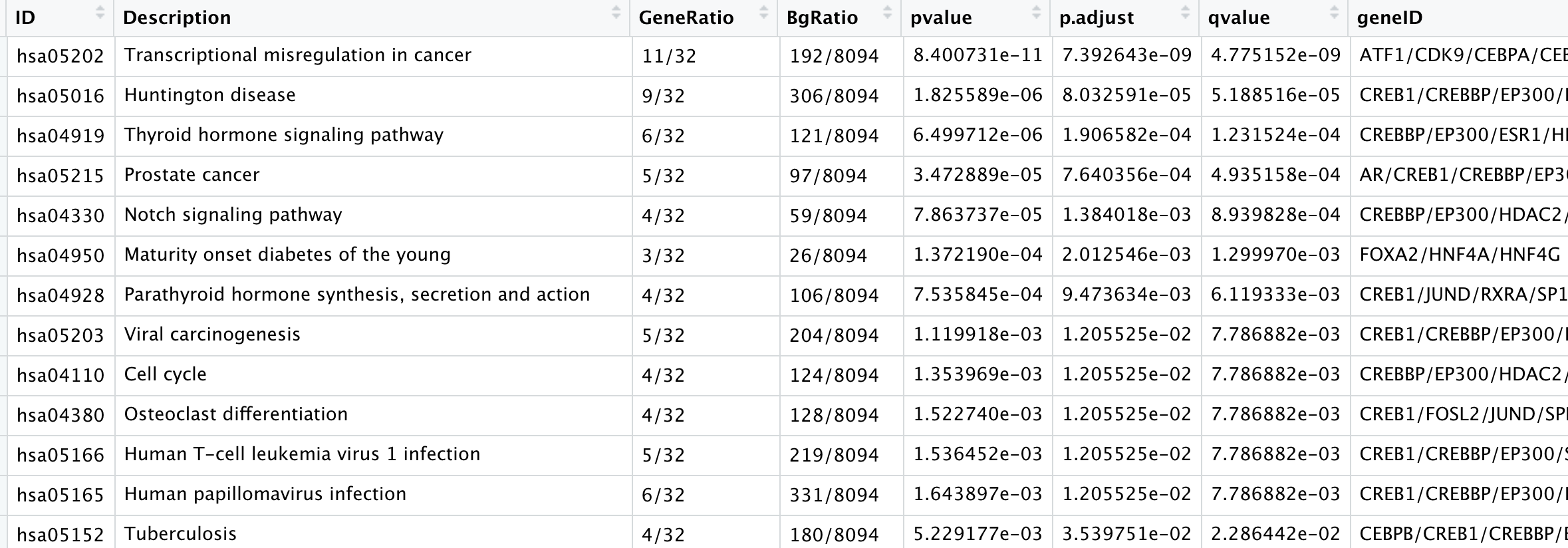
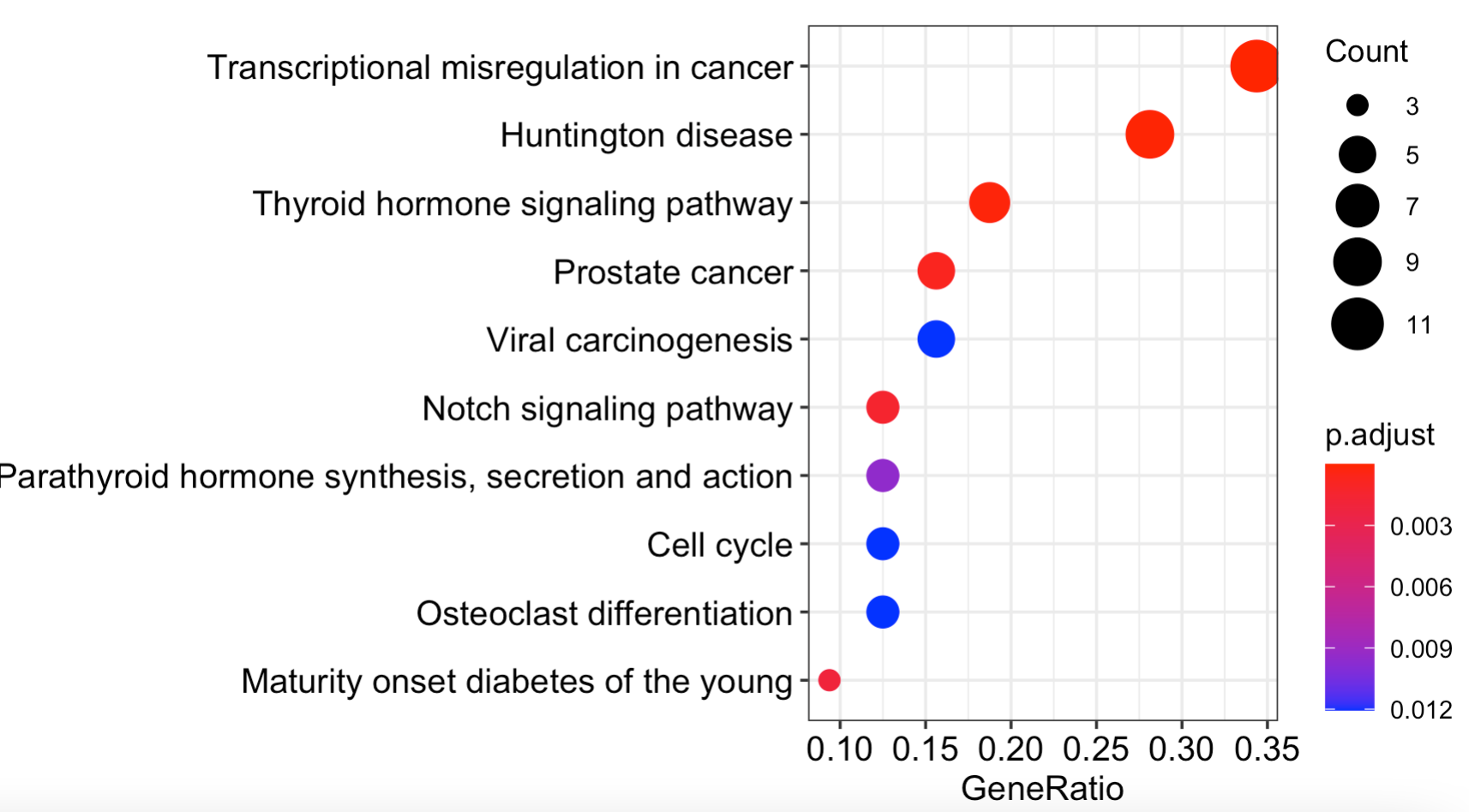
把上面的表格内容,进行可视化如下:
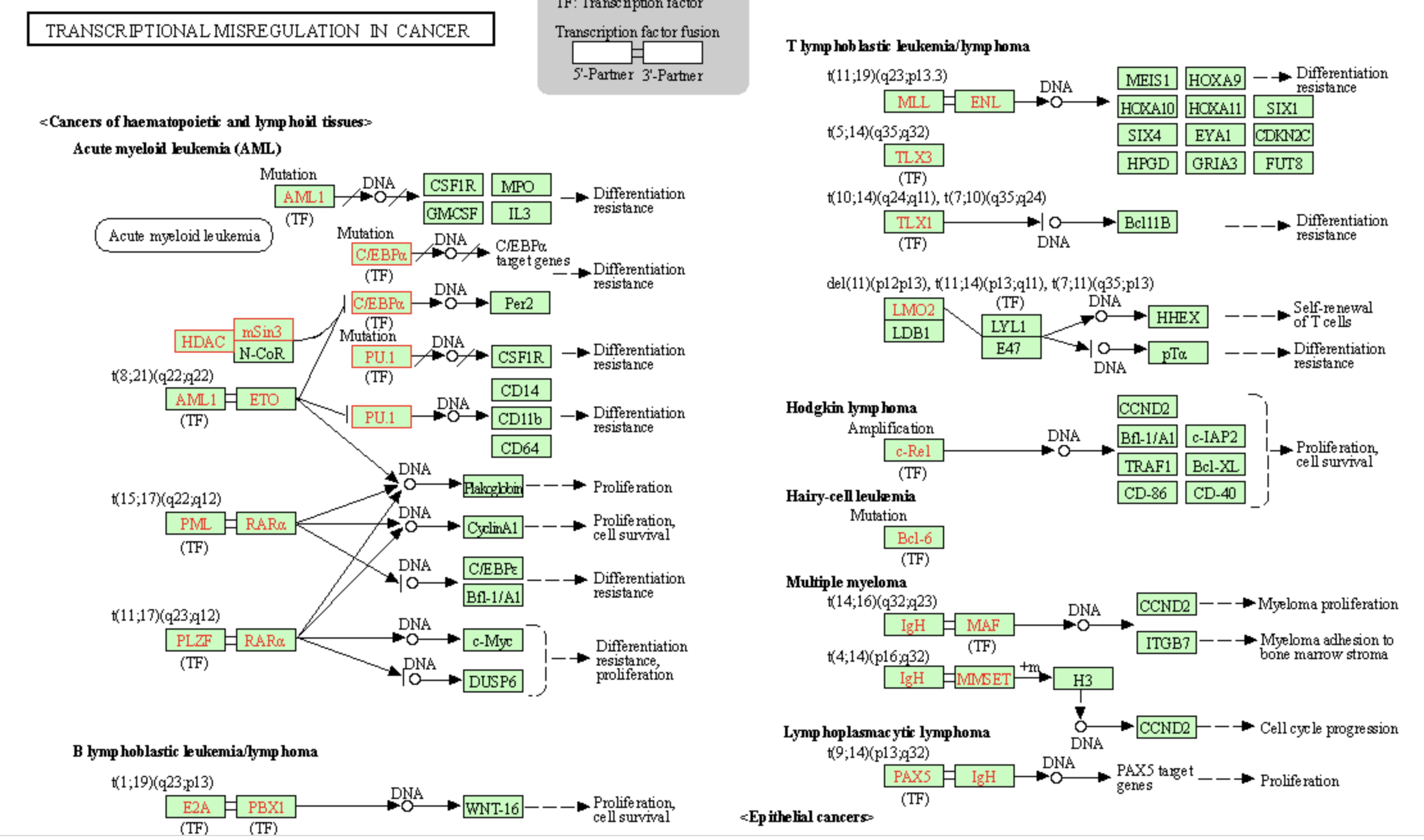
如果感兴趣具体的通路,可以使用 browseKEGG(enrichKK, ‘hsa03430’) 去自己的电脑的浏览器可视化:
这个函数其实就是帮我们拼接好了一个url,输入浏览器,这个url是:
我们富集拿到的这个通路就被放大的一清二楚:
第三步:看GO数据库注释
代码如下:
go <- enrichGO(gene_up, OrgDb = "org.Hs.eg.db", ont="all",
pvalueCutoff = 0.9999,qvalueCutoff = 0.999999)
library(ggplot2)
library(stringr)
barplot(go, split="ONTOLOGY")+ facet_grid(ONTOLOGY~., scale="free")
barplot(go, split="ONTOLOGY",font.size =10)+
facet_grid(ONTOLOGY~., scale="free") +
scale_x_discrete(labels=function(x) str_wrap(x, width=50))+
ggsave('gene_sur_GO_all_barplot.png')
go=DOSE::setReadable(go, OrgDb='org.Hs.eg.db',
keyType='ENTREZID')
tmp=go@result
GO数据库分成了,BP,CC,MF ,它们富集结果表格进行条形图可视化如下:
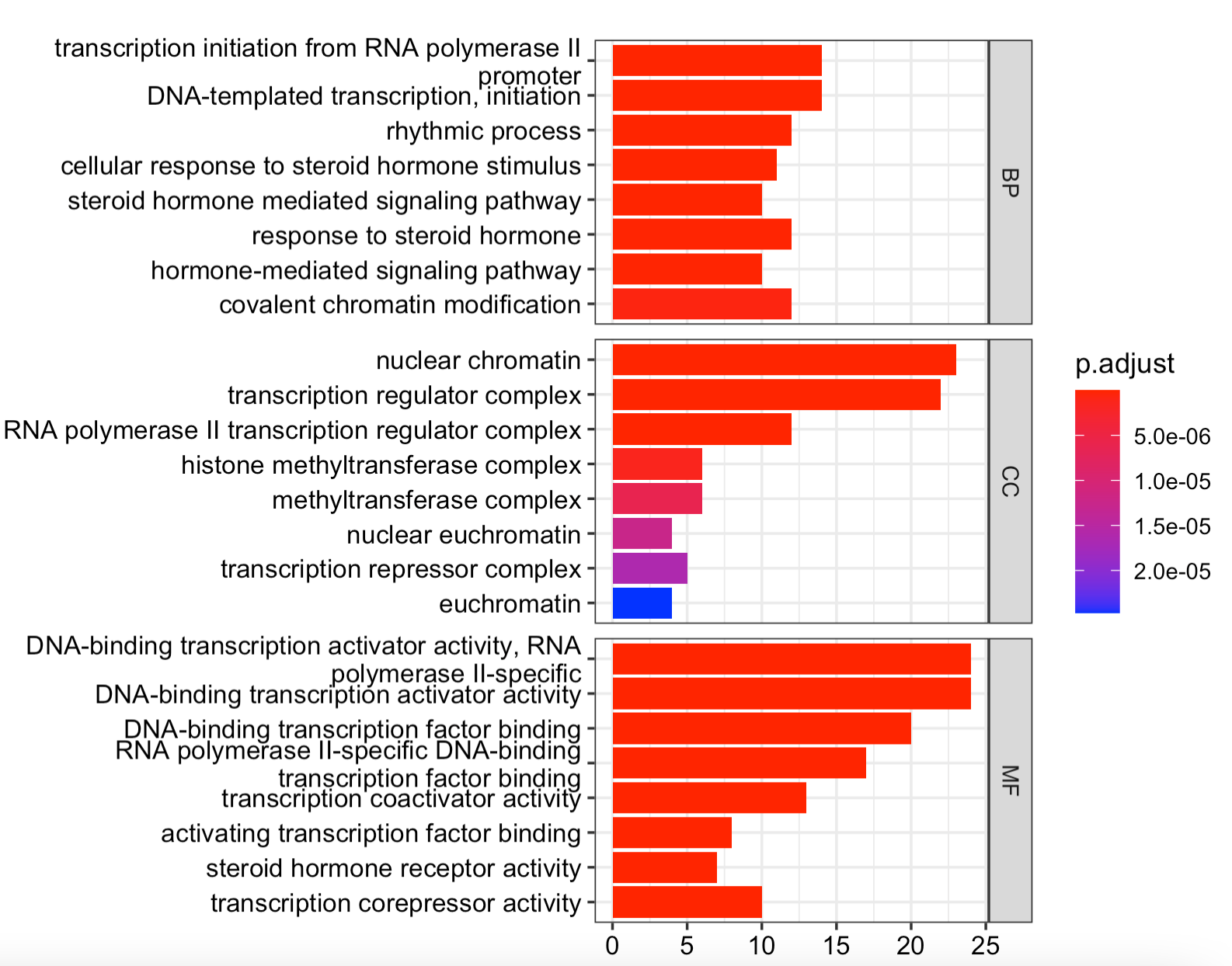
如果感兴趣表格内容,也可以把 tmp=go@result 输出成为csv文件慢慢看。
第四步:高级可视化
主要是针对前面的KEGG数据库结果举例,代码是:
cnetplot(enrichKK, categorySize="pvalue", colorEdge = TRUE)
cnetplot(enrichKK, circular = TRUE, colorEdge = TRUE)
emapplot(enrichKK)
heatplot(enrichKK )
这4个函数的效果如下:
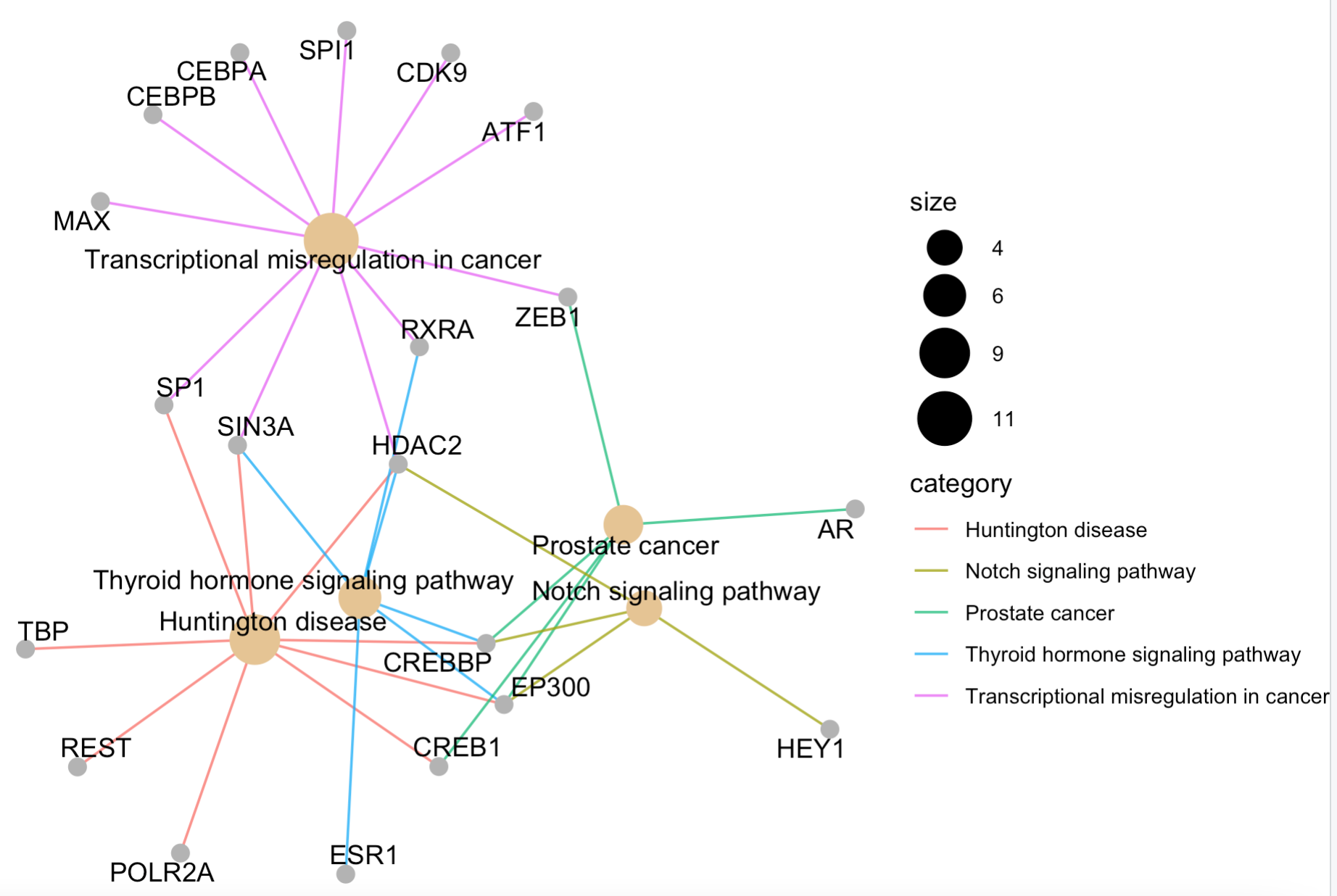
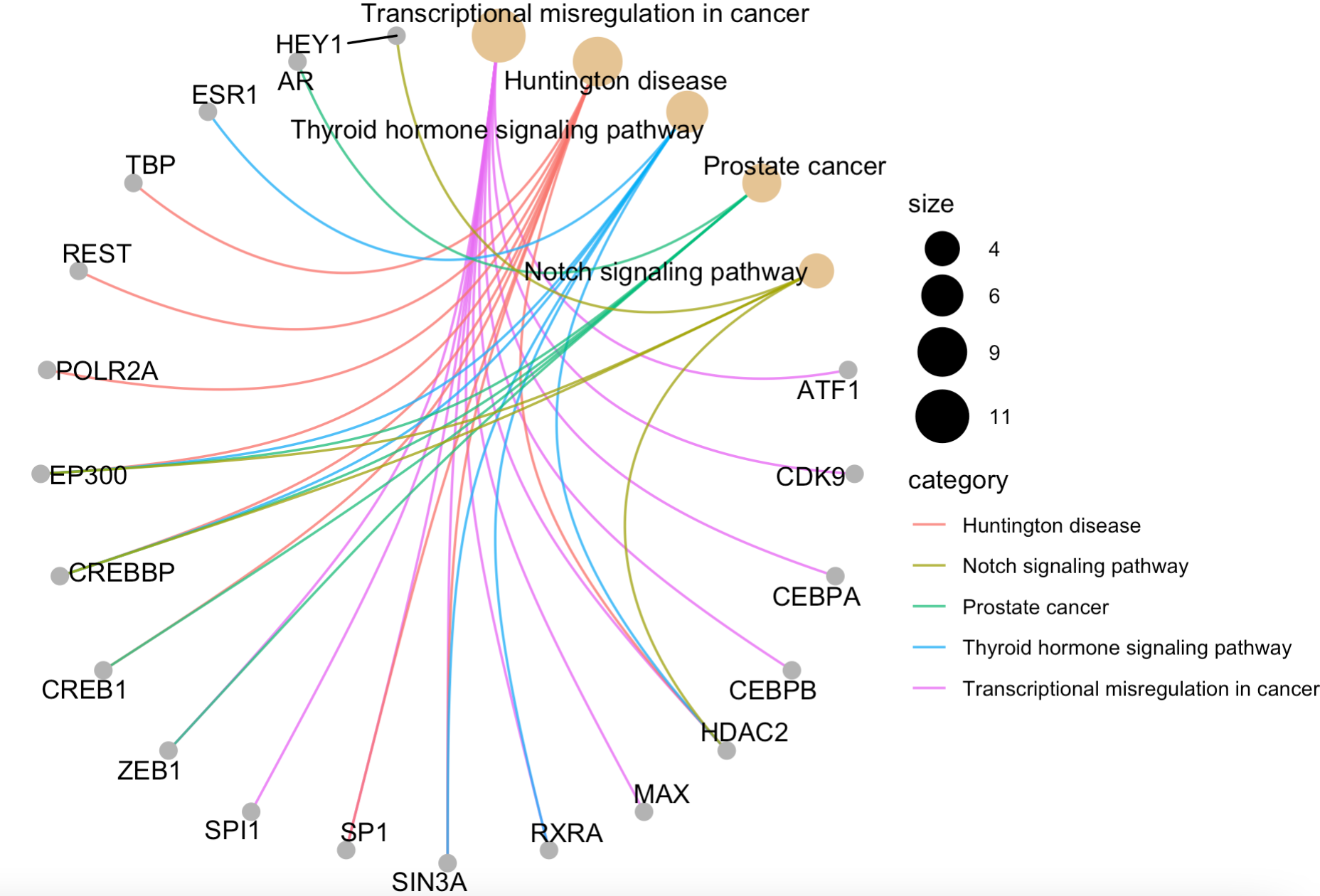
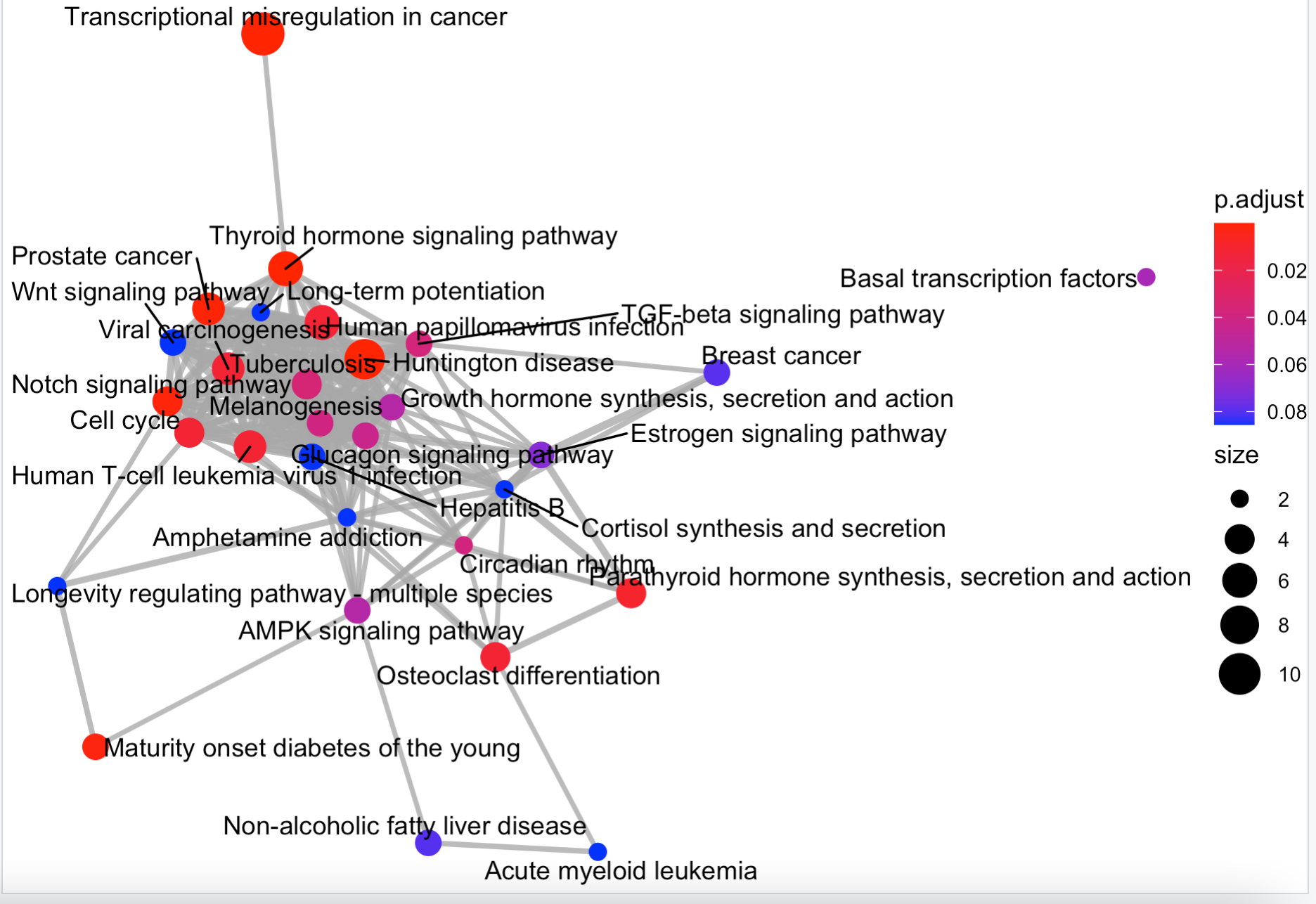
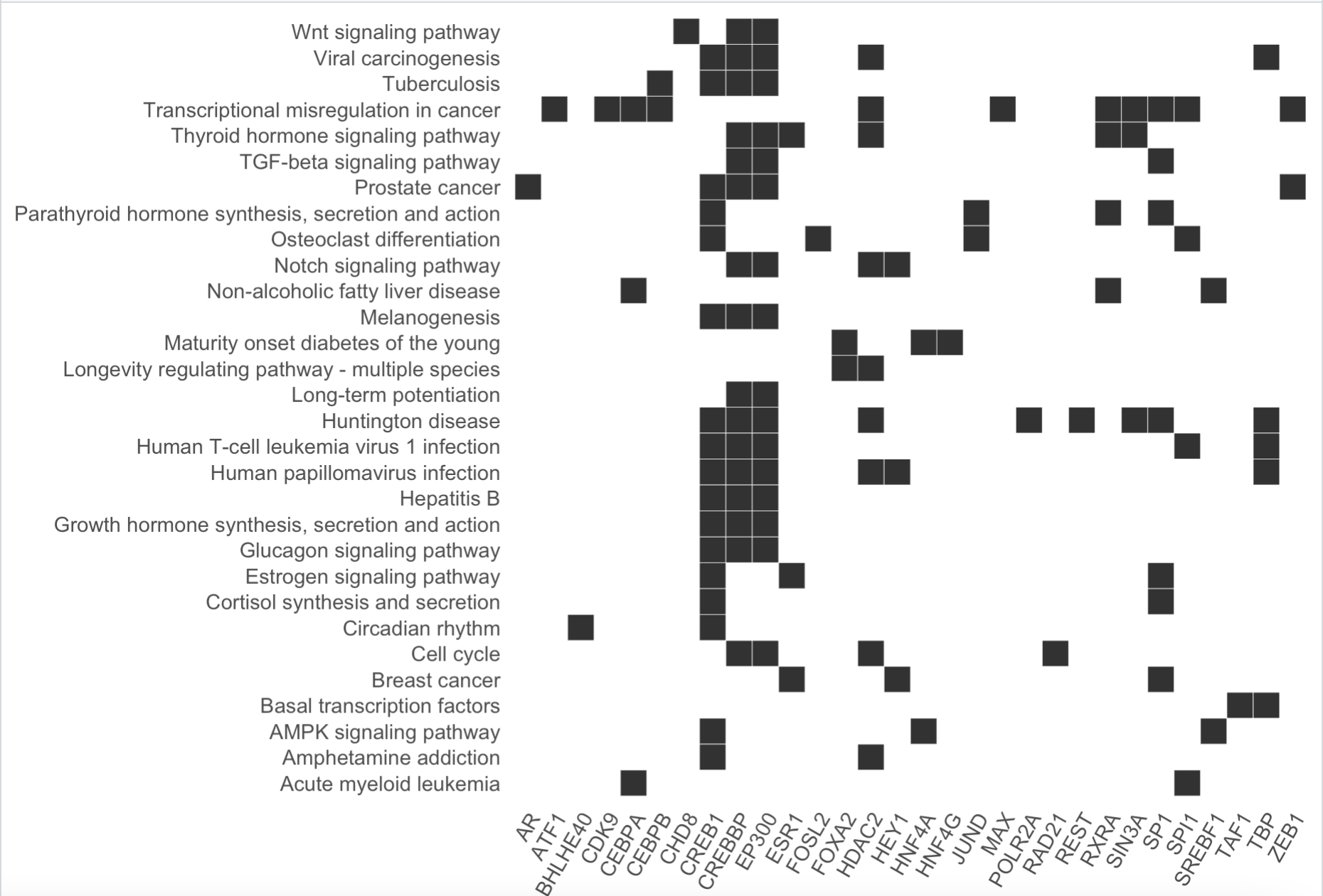
那么问题来了
开局的50个基因是如何得到的呢?
首先可以是差异分析的上下调基因,然后可以是生存分析的统计学显著的预后基因,甚至更多。
如果是差异分析,基本上看我五年前的《数据挖掘》系列推文 就足够了;