我看到2020年10月发表在cancer cell杂志的文章《Senescence Reprogramming by TIMP1 Deficiency Promotes Prostate Cancer Metastasis》,链接是:https://doi.org/10.1016/j.ccell.2020.10.012 ,该研究里面有一个bulk转录组数据和一个单细胞转录组数据,槽点太多了,我们一个个来吐。首先转录组数据在:https://www.ebi.ac.uk/ena/browser/view/PRJEB40610 ,是RNA seq of prostate gland from WT, PTEN pc -/- and PTEN pc-/-;P53 -/- mice , 3*3=9 的样品,超级简单的一个实验设计啦。
让我们一起来看看该研究的文献标题里面的TIMP1 基因是如何得到的吧,下面的一个 Cytokine and cytokine-related genes 的表达量热图,如下所示:
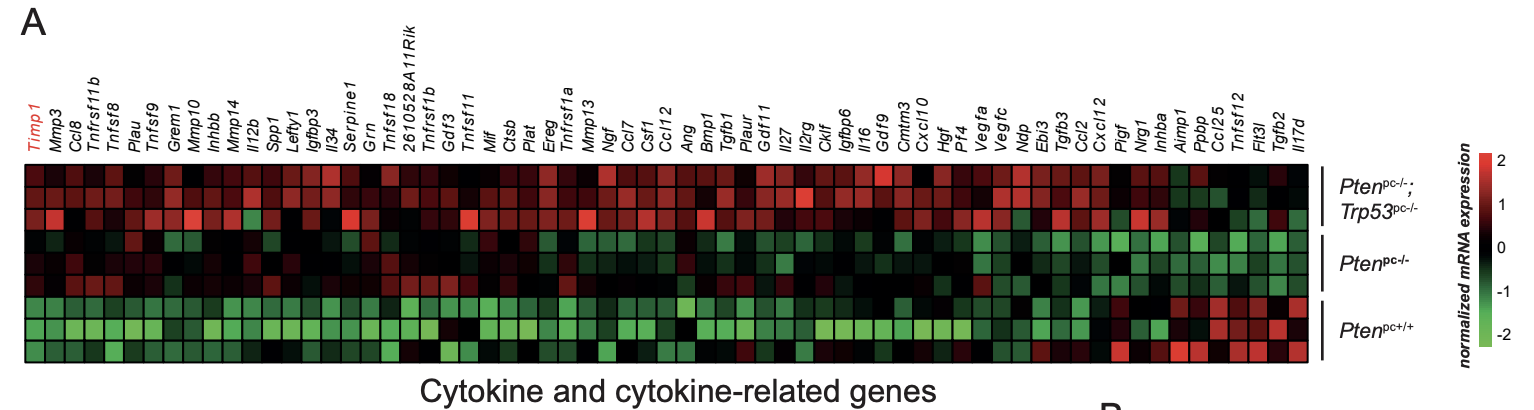
文章里面描述的是这个TIMP1 基因差异最大,真的是让人笑掉大牙,全局筛选怎么可能会有最大值?就算是有最大值,那么它和第二大你凭什么说最大就最有意思?
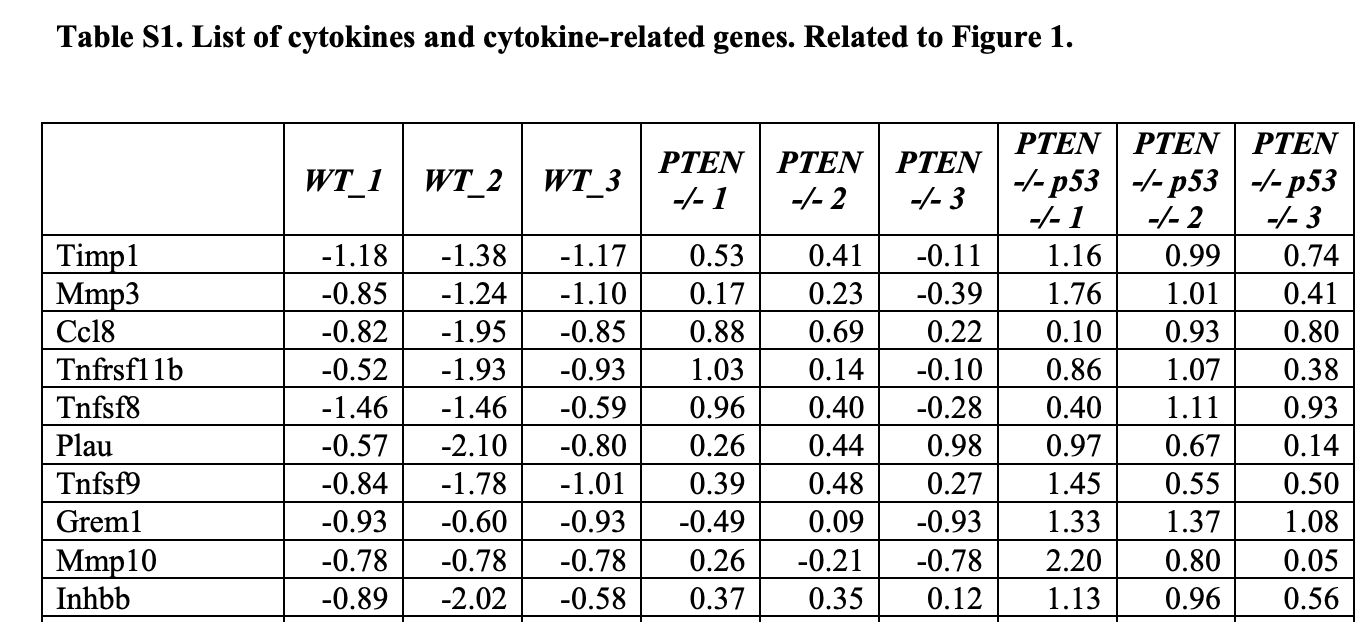
而且,你看看作者列出来的表达量矩阵??你告诉我这个是RNA-seq的结果???怕不是本科生瞎编的吧!!!
然后关注了 senescence-associated secretory phenotype (SASP) ,也是如下所示的热图

既然如此为什么不关注 matrix metalloproteinases (MMPs)呢 ?
你们这些人,做单细胞转录组或者转录组完全是瞎搞,为了蹭我们NGS的热点, 没有认真的学习RNA-seq数据的特性,仅仅是凑数!
我鄙视你们!