前面我们已经完成了cytof数据处理的主要步骤,读入文件,质量控制,降维聚类分群,生物学注释和细胞亚群比例差异分析。 目录如下:
- 1.cytof数据资源介绍(文末有交流群)
- 2.cytofWorkflow之读入FCS文件(一)
- 3.cytofWorkflow之构建SingleCellExperiment对象(二)
- 4.cytofWorkflow之基本质量控制(三)
- 5.cytofWorkflow之聚类分群(四)
- 6.cytofWorkflow之人工注释生物学亚群(五)
- 7.cytofWorkflow之亚群比例差异分析(六)
其实跟纯粹的单细胞转录组就非常类似了,不过单细胞转录组数据分析的细节以及背景我就不赘述了,看我在《单细胞天地》的单细胞基础10讲:
- 01. 上游分析流程
- 02.课题多少个样品,测序数据量如何
- 03. 过滤不合格细胞和基因(数据质控很重要)
- 04. 过滤线粒体核糖体基因
- 05. 去除细胞效应和基因效应
- 06.单细胞转录组数据的降维聚类分群
- 07.单细胞转录组数据处理之细胞亚群注释
- 08.把拿到的亚群进行更细致的分群
- 09.单细胞转录组数据处理之细胞亚群比例比较
以及各式各样的个性化汇总教程,差不多就明白了。
基本流程走完了,仅仅是万里长征第一步而已。我们可以开始尝试分析一些文献的公共数据集啦,不过在处理那些数据的过程中,我们还需要传授给大家几个小技巧。
首先取出亚群
前面的细胞亚群生物学命名的时候,我们就讲解过,根据指定抗体信号强度,可以取子集,代码如下:
rm(list = ls())
suppressPackageStartupMessages(library(flowCore))
suppressPackageStartupMessages(library(diffcyt))
suppressPackageStartupMessages(library(HDCytoData))
require(cytofWorkflow)
load(file = 'K20_output_of_cytofWorkflow.Rdata')
sce@metadata
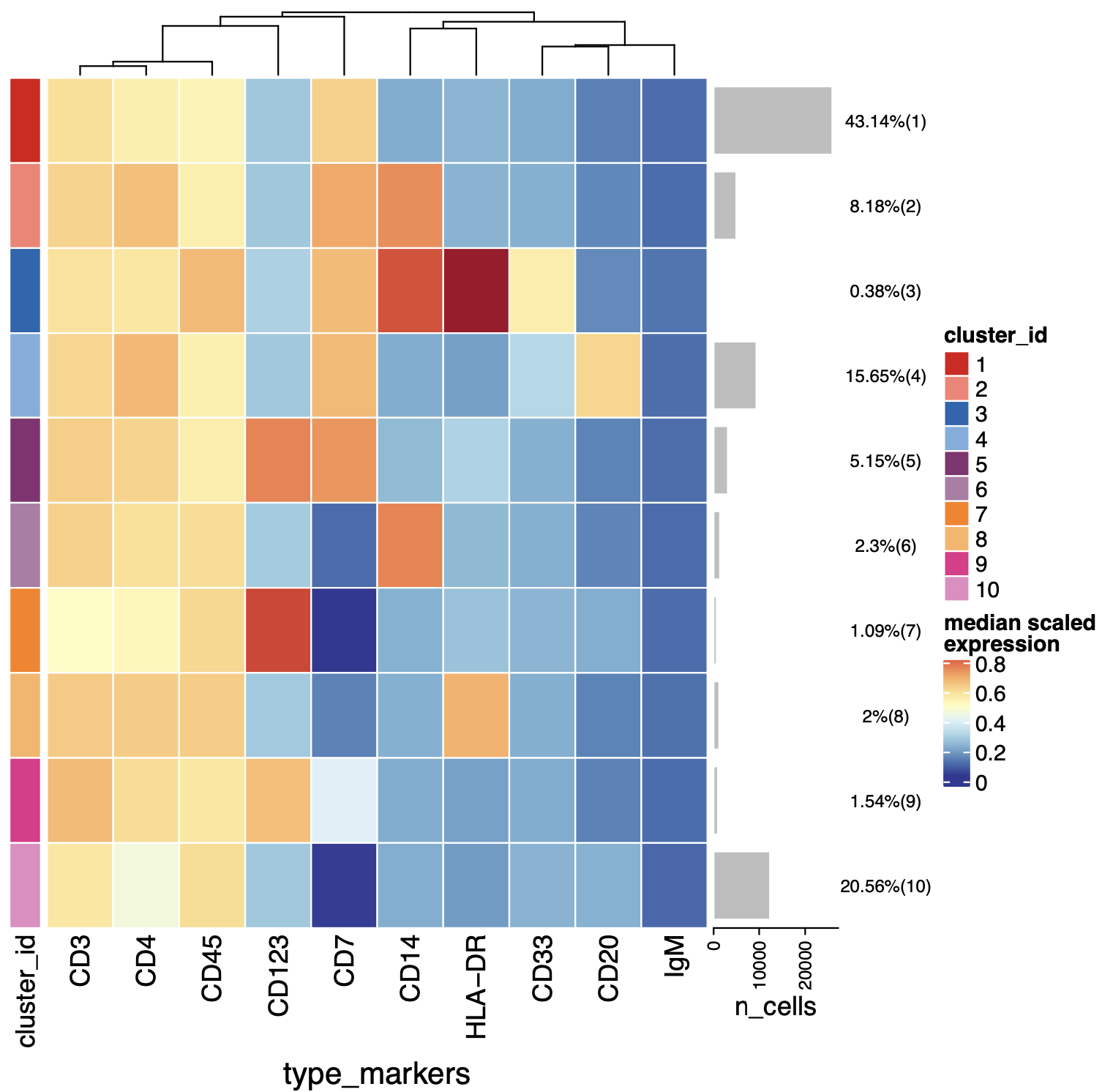
p=plotExprHeatmap(sce, features = "type",
by = "cluster_id", k = "meta20",
row_clust = F,
bars = TRUE, perc = TRUE)
p
p@matrix
kp=p@matrix > 0.5
colnames(kp)
k1=which(kp[,"CD45"] & kp[,"CD3"] & kp[,"CD4"] );k1 # CD4
sce
som=sce@metadata$cluster_codes
table(colData(sce)$cluster_id)
choose_som=som[som$meta20 %in% k1,1]
choose_som
这样取出来了就是可能的CD4的T细胞啦,有了这个choose_som后面就可以获取。
不知道大家是否还记得Seurat分析流程里面的单细胞对象取子集呢?其实本质上都是考验大家对R里面的S4对象的理解而已。
然后重新组合成为新的SingleCellExperiment对象
SingleCellExperiment对象的认识非常重要,重点就是:
- different quantifications (e.g., counts, CPMs, log-expression) can be stored simultaneously in the assays slot,
- row and column metadata can be attached using rowData and colData, respectively.
全部的代码如下;
sce
ct=assay(sce,i = 1)[,colData(sce)$cluster_id %in% choose_som]
ex=assay(sce,i = 2)[,colData(sce)$cluster_id %in% choose_som]
phe=colData(sce)[colData(sce)$cluster_id %in% choose_som,]
geneinfor=rowData(sce)
sce <- SingleCellExperiment(list(counts=ct,exprs=ex),
colData=phe,
rowData=geneinfor
)
sce
新组建的SingleCellExperiment对象就可以继续走聚类分群的步骤啦。而且很容易建议,代码如下:
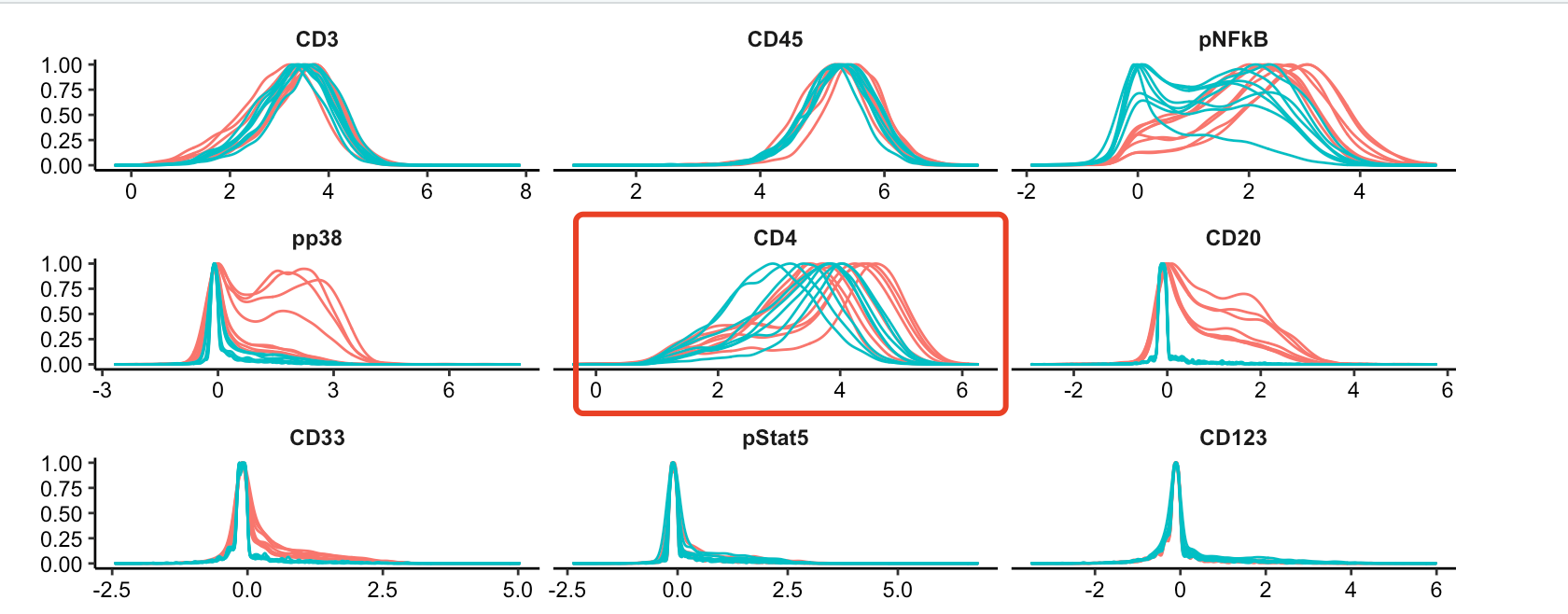
p <- plotExprs(sce, color_by = "condition")
p$facet$params$ncol <- 3
p
可以看到,CD4都是有表达量的:
同样的聚类分群代
跟之前一样,示例代码如下:
sce
pro='Cd4-Tcells_sub'
p <- plotExprs(sce, color_by = "condition")
p$facet$params$ncol <- 3
p
ggsave2(filename = paste0(pro,'_all_markers_density.pdf'),height = 11)
n_cells(sce)
plotCounts(sce, group_by = "sample_id", color_by = "condition")
ggsave2(filename = paste0(pro,'_plotCounts.pdf'))
# 10 lineage markers and 14 functional markers across all cells measured for each sample in the PBMC dataset
pdf( paste0(pro,'_plotExprHeatmap.pdf'))
plotExprHeatmap(sce, scale = "last",
hm_pal = rev(hcl.colors(10, "YlGnBu")))
dev.off()
# PCA-based non-redundancy score (NRS)
plotNRS(sce, features = "type", color_by = "condition")
ggsave2(filename = paste0(pro,'_plotNRS.pdf'))
set.seed(1234)
# 通常建议第一次分群,细一点要好,后面可以人工合并
# We call ConsensusClusterPlus() with maximum number of clusters maxK = 20.
sce <- cluster(sce, features = "type",
xdim = 10, ydim = 10, maxK = 10, seed = 1234)
pdf(paste0(pro,'_cluster_plotExprHeatmap_row_clust_F.pdf'))
plotExprHeatmap(sce, features = "type",
by = "cluster_id", k = "meta10",
row_clust = F,
bars = TRUE, perc = TRUE)
dev.off()
pdf(paste0(pro,'_plotClusterExprs.pdf'))
plotClusterExprs(sce, k = "meta10", features = "type")
dev.off()
pdf(paste0(pro,'_cluster_plotMultiHeatmap.pdf'))
plotMultiHeatmap(sce,
hm1 = "type", k = "meta10",
row_anno = FALSE, bars = TRUE, perc = TRUE)
dev.off()
# 节约计算资源
# run t-SNE/UMAP on at most 500/1000 cells per sample
set.seed(1234)
sce <- runDR(sce, "TSNE", cells = 1e4, features = "type")
sce <- runDR(sce, "UMAP", cells = 1e4, features = "type")
# plotDR(sce, "UMAP", color_by = "CD4")
library(ggplot2)
p1 <- plotDR(sce, "TSNE", color_by = "meta10") +
theme(legend.position = "none")
p2 <- plotDR(sce, "UMAP", color_by = "meta10")
lgd <- get_legend(p2)
p2 <- p2 + theme(legend.position = "none")
plot_grid(p1, p2, lgd, nrow = 1, rel_widths = c(5, 5, 2))
ggsave2(filename = paste0(pro,'_umap_vs_tSNE.pdf'))
# facet by sample
plotDR(sce, "TSNE", color_by = "meta10", facet_by = "sample_id")
ggsave2(filename = paste0(pro,'_TSNE_by_samples.pdf'))
# facet by condition
plotDR(sce, "TSNE", color_by = "meta10", facet_by = "condition")
ggsave2(filename = paste0(pro,'_TSNE_by_condition.pdf'))
# facet by sample
plotDR(sce, "UMAP", color_by = "meta10", facet_by = "sample_id")
ggsave2(filename = paste0(pro,'_umap_by_samples.pdf'))
# facet by condition
plotDR(sce, "UMAP", color_by = "meta10", facet_by = "condition")
ggsave2(filename = paste0(pro,'_umap_by_condition.pdf'))
plotCodes(sce, k = "meta10")
pdf(paste0(pro,'_cluster_som100_plotMultiHeatmap.pdf'))
plotMultiHeatmap(sce,
hm1 = "type", k = "som100", m = "meta10",
row_anno = FALSE, col_anno = FALSE, bars = TRUE, perc = TRUE)
dev.off()
sce@metadata
plot_grid(labels = c("A", "B"),
plotDR(sce, "UMAP", color_by = "meta10"),
plotDR(sce, "UMAP", color_by = "meta8"))
plotAbundances(sce, k = "meta10", by = "sample_id")
ggsave2(filename = paste0(pro,'_plotAbundances_barplot.pdf'))
plotAbundances(sce, k = "meta10", by = "cluster_id", shape_by = "patient_id")
ggsave2(filename = paste0(pro,'_plotAbundances_boxplot.pdf'))
save(sce,file = paste0(pro,'k10_output_of_cytofWorkflow.Rdata'))
可以看到初步分成的10个群,确实都有不一样的地方,如果生物学背景足够,就可以给出解释。