翻译自:https://constantamateur.github.io/2020-10-24-scBatch2/
更多教程见其博客:https://constantamateur.github.io/
Part 2 - 不用矫正的方法来处理批次效应
2020/10/24
在上一篇文章中,作者曾写过过度处理批次效应的后果。这里可能会有些争议,他将对如何在不使用不知道具体原理的“批次效应矫正”工具的情况下处理批次效应给与一些建议。为什么要这么做?批次效应矫正工具真的那么糟糕吗?
并不是作者认为批次效应矫正方法不好。而是它们并不能百分百清楚的告诉用户到底怎么处理了数据,处理到什么程度。因此,先自己处理数据,如果批次效应实在是很影响后续分析,再来使用所谓的批次矫正,因为并不是所有的批次效应都可以或者应该被矫正。
我们的公众号生信技能树之前也写了很多关于批次效应的推文,例如
- 并不是所有的批次效应都可以被矫正
- 关于批次效应矫正后出现负值
- 校正批次效应
- GSE83521/GSE89143数据集-需去除批次效应
- PCA图显示分组无差异,怎么办?
大家可以当做是背景知识读一下!去除已知的批次效应
哈哈哈作者这句话很有意思,我的数据被诅咒了所以不好分析..其实是可能有一些批次效应作祟。
批次效应主要是想要去除实验不同步骤之间的系统差异。需要保持实验中的变量除外的环境都一致,这样才会突出变量的差异。有些是生物学差异,但最主要的差异是由技术效应导致的。最常见的情况是,批次效应由许多大大小小的差异导致,每个差异都造成了总体不必要的差异的一部分。
也许是我们对不同的样品使用了不同批次的试剂、用过不同化学成分的试剂盒、完全不同的单细胞测序平台、一个样本必须在冰上保存一夜、改变了组织分离方法、不同的样品在不同的测序平台。这些改变中的每一个都可能导致不同的结果。
虽然其中一些差异将导致很难甚至不可能预测和逆转的转录变化,还有一些则可以轻易去除。因此,对有批次效应的数据,可以做的事就是先去除已知的批次效应。
这里将重点介绍 10X 单细胞表达数据。去除 swapped barcodes
如图所示,在新型号的 Illumina 测序机上,流动池中的连接和DNA扩增同时进行,各步骤之间不需要对流动池进行任何清洗。样本标记仍保留在溶液中,并且可以使用具有不同标记的库中的DNA分子作为模板扩展。错误标记的分子在流动池的纳米孔之间的转移导致错误标记的DNA分子的聚集和测序。
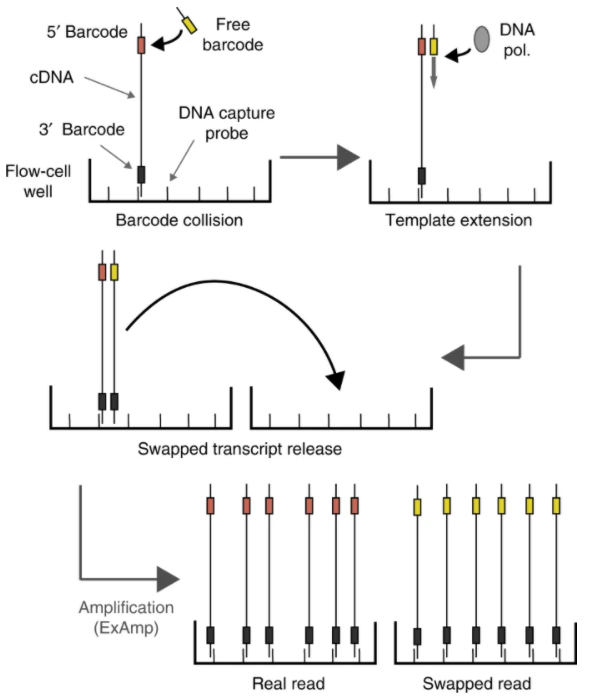
首先去除 swapped barcodes. 用这个R包 DropletUtils 来处理很简单~ (作者给与了友情提示说这个包不好下载,结果嗖的一下就下完了..eee).
library(DropletUtils) cleaned = swappedDrops('data/outs/molecule_info.h5')大多数情况下,这一步并不能起到多大作用。但曾经有过这样的情况,在新型的以Nano-Well 为特点的Patterned Flow Cell Technology(PFCT)的测序仪上运行的数据得到了极大的改善。在较新的 10X kits 中引入双重标签应该可以避免这个问题。
去除环境RNA
Ambient gene expression(环境基因表达)是指不是来自 barcode 细胞,而是来自其他溶解细胞的count,这些细胞的 mRNA 在文库构建之前污染了细胞悬液。这些增加的环境计数会扭曲下游分析,如标记基因鉴定或其他差异表达检测,尤其是当样本之间的水平变化时。在基于液滴的 scRNA-seq 数据集中校正这些影响是可能的,由于大量的空液滴,可用于模拟环境RNA表达谱。SoupX使用这种方法直接纠正计数数据。
第二件事是去除环境RNA污染。这一点非常重要,因为环境RNA看起来与实验中所有细胞的平均值非常相似。下图显示了这一点,它将背景污染中每个基因的平均表达与实验中所有细胞的平均表达进行了比较。
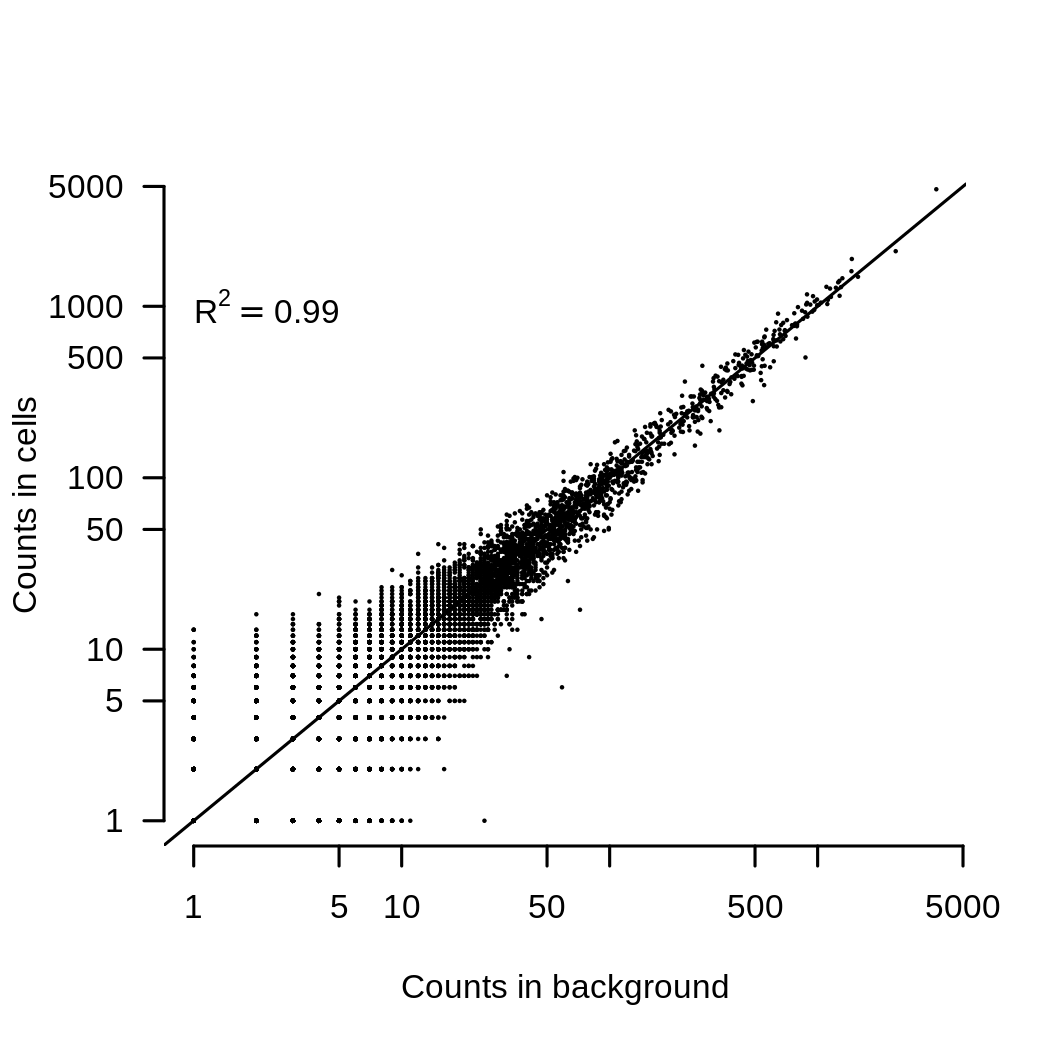
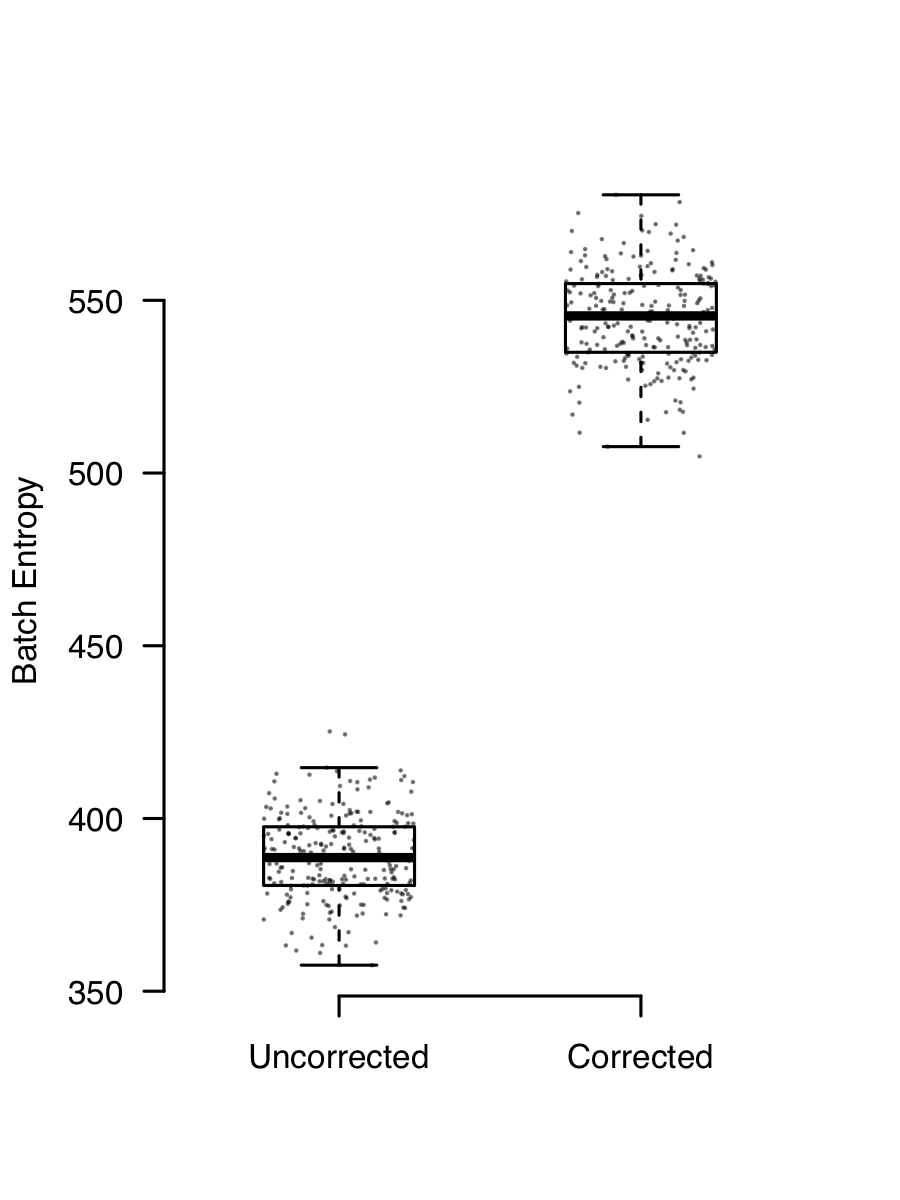
这样做的结果是,如果有两个(或更多)具有不同混合细胞的数据,最终看起来会有所不同,因为背景环境不同。去除环境污染可以使用作者写的这个 SoupX ,但也有其他方法。环境RNA污染会产生一种几乎总是存在的批次效应,去除它可以明显提高整合度。此图显示了将 SoupX 应用于某些数据之前和之后的批处理熵。熵越高,表明去除背景时混合效果越好。
在这篇论文 SoupX paper 里对环境RNA污染及其影响有更完整的解释。处理细胞周期
细胞处于细胞周期的不同阶段并不是严格意义上的批次效应。但是它的关联性足够强,并且可以对数据聚类的方式产生很大影响,所以这里还是要讨论一下。周期细胞可以使真正不相关的细胞看起来是相关的,因为它们都处于 S、G2 或 M 期。大多数人对作者的担忧的回应都是这样的:“没关系,可以回归的去除细胞周期的影响。”
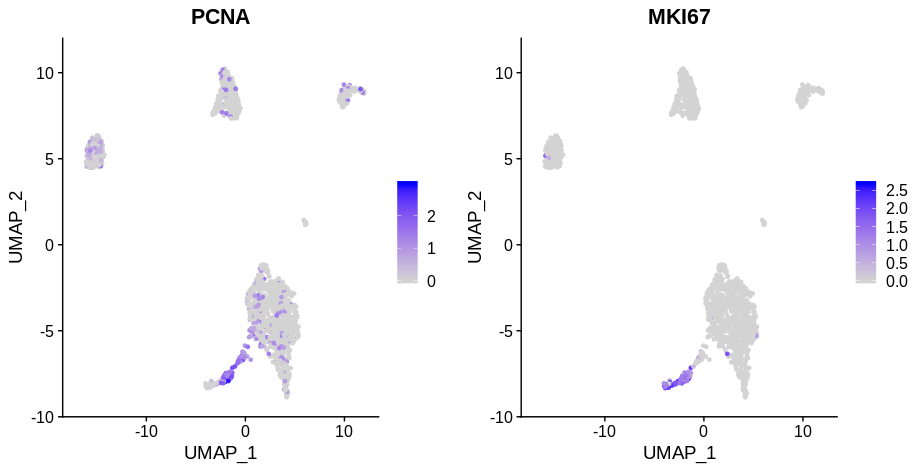
下面的图表可以清楚地看到,处于 S 期( PCNA 阳性)和 G2M 期( MKI67 期)的细胞与它们来自哪个簇都是不同的(这些细胞是 this paper 中的乳头状肾细胞癌细胞)。在这里,使用 PCNA 作为 S 期标记物,MKI67 作为 G2M 期标记物,作者认为观察标记物的表达比 Seurat 的 CellCycleScoring 函数更可靠。(这就是生物学背景知识了,在大家处理数据的过程中要尽可能多记忆一些重要的细节知识点!!!)
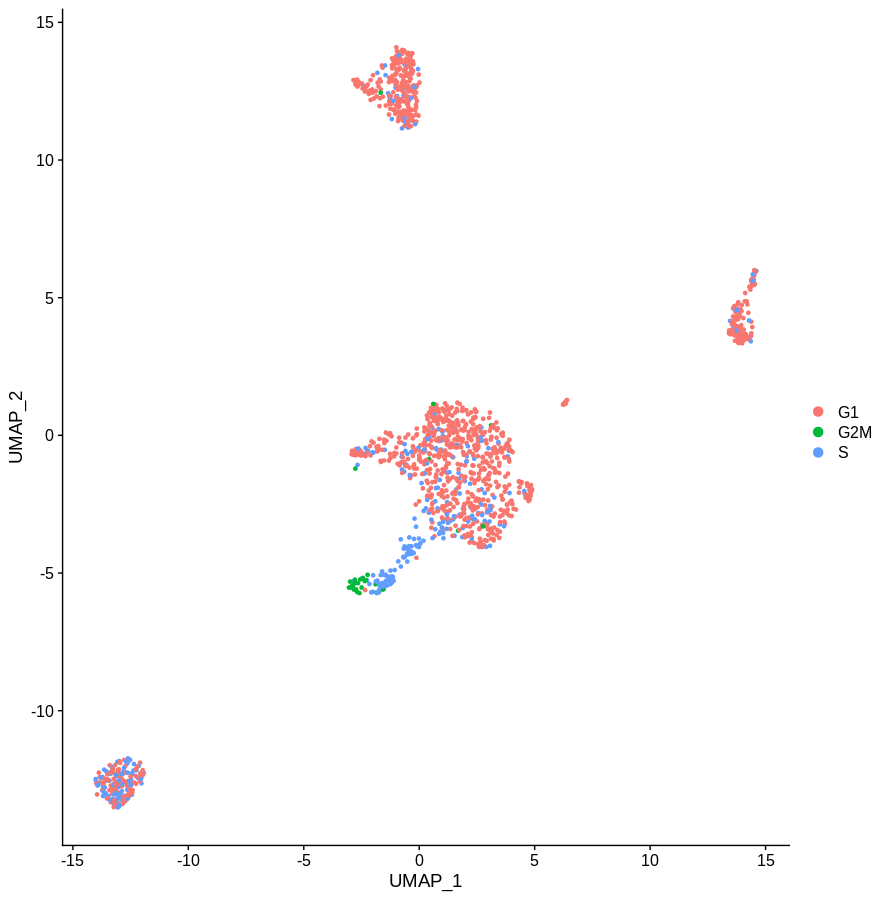
去除细胞周期基因
但是如果我们丢弃所有的周期基因呢?这应该会使它们和 G1 期细胞一样吧?在Seurat我们可以很容易地做到这一点
phaseS = cnts['PCNA',]>0
phaseM = cnts['MKI67',]>0
altCnts = cnts[!rownames(cnts) %in% unlist(cc.genes.updated.2019),]
#Usual stuff...
altCnts@meta.data$Phase = ifelse(phaseS,'S',ifelse(phaseM,'G2M','G1'))
DimPlot(altCnts,group.by='Phase')
显然,处于周期的哪个阶段的定义有点粗略,但概念相当清晰。去除主要的细胞周期基因并没有起到多大作用。如果你能找到一份更完整的细胞周期相关基因列表,应该会更好的去除这些基因。但是,考虑到基因在不同环境中具有不同的功能,这种方法发挥作用的几率很小。(生物学知识确定有点杂乱无章)
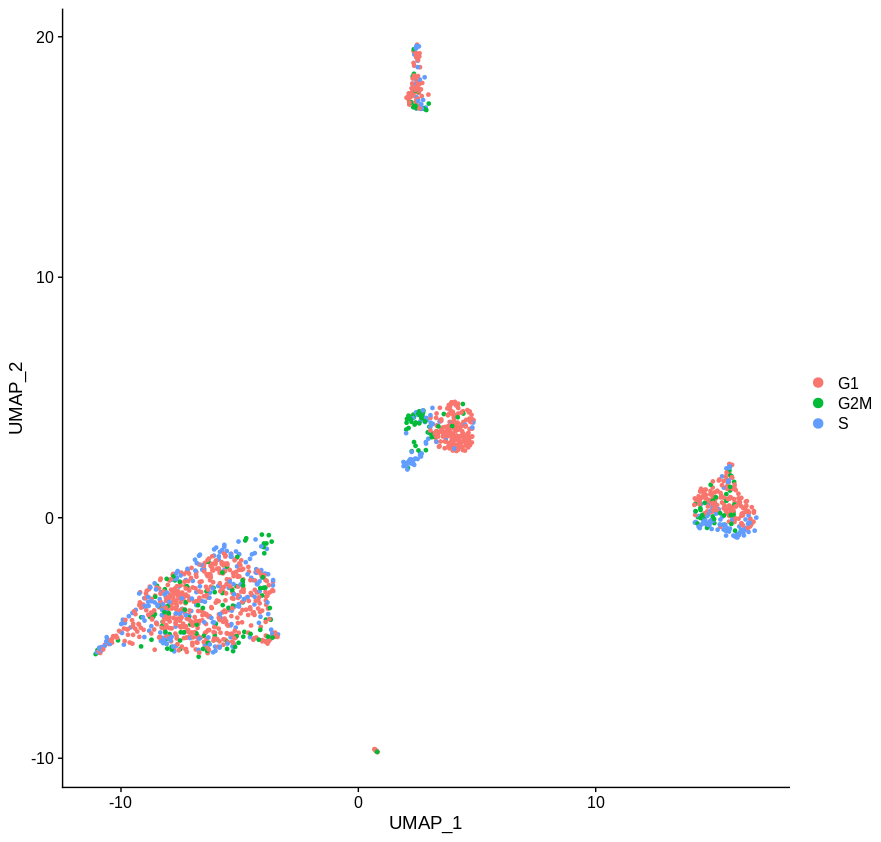
回归的去除细胞周期的影响
使用 Seurat vignette 来看看 regress cell cycle out 会怎样:
reg = CreateSeuratObject(cnts)
reg = NormalizeData(reg)
reg = CellCycleScoring(reg,s.features=cc.genes.updated.2019$s.genes,g2m.features=cc.genes.updated.2019$g2m.genes)
reg = ScaleData(reg,vars.to.regress=c('S.Score','G2M.Score'))
reg = FindVariableFeatures(reg)
reg = RunPCA(reg)
reg = RunUMAP(reg,dims=seq(50))
DimPlot(reg,group.by='Phase')
这看起来好多了,但显然还是有区别的。也许这种差异是潜在的细胞状态之间的某种真正的差异,这种状态不是细胞周期,而是细胞本身固有的。但是,除非你真的缺乏细胞,或者有一些细胞类型只存在于S/G2M期,否则根本不需要这么麻烦,只需去除周期细胞,这就不再是问题了。
去除无关紧要的基因
在某些情况下,在分析之前去除不需要的基因可以有效防止批次效应的产生。
例如,有一些已知的基因会因组织处理而发生变化(例如,热休克基因)。在这种情况下,你很可能会怀疑和这些基因相关的一些簇是否是和基因相关,或者是实验处理过程中的变化。那么为什么不干脆把他们排除在外呢?要排除的基因通常是线粒体基因和热休克基因。但这还取决于实验设计。
显然,这些基因中的信息有时仍然有用,这个想法只是为了防止它们聚类。可能最稳健的方法是将它们从输入计数矩阵中排除,但将信息作为元数据保留,以便不时之需。例如,
mtGenes=grep('^MT-',rownames(cnts),value=TRUE)
redCnts = cnts[!rownames(cnts) %in% mtGenes,]
mtFrac = colSums(cnts[mtGenes,])/colSums(cnts)
red = CreateSeuratObject(redCnts)
red@meta.data$mtFrac = mtFrac
这将消除MT基因对分析的任何影响,但是如果您希望使用作为元数据提供的信息。另一种选择是将它们从用于计算PC和下游分析的高度可变基因中排除。例如。
reg@assays$RNA@var.features = setdiff(reg@assays$RNA@var.features,mtGenes)
其实处理到最后,会感觉我们重要的单细胞转录组其实并不是全局的基因表达量啦,有点类似于特定的gene panel,也就是说目前主流的10X单细胞仪器其实可以被BD平台的单细胞取代?
管它干啥~
批次效应在 tSNE 或 UMAP 上看起来很难看,可能至少会有一个编辑抱怨它。
主要取决于想要在分析中实现什么目标,以及通过改变数据来消除批次效应是否真的会进一步推动这一目标。
例如,单细胞分析的一个共同目标是在实验中定义特定细胞类型特有的基因。从表面上看,将你的细胞类型分成多个簇似乎会阻碍这一努力。但是,所有这些实际上都会使注释数据的任务变得更加困难。作者从来没有遇到过不想合并两个按算法选择的簇的注释的单细胞分析。
虽然没有任何确凿的证据,但作者认为如果提供的数据没有通过批次效应处理,那么标记查找方法将会做得和处理后的数据一样好,甚至更好。当然,如果您将标记查找定义为一项差异表达任务 (which I don’t think you should),那么使用 edgeR 或 DESeq2 会得到更好的结果,这些方法需要输入 raw counts。
结论
有时,你的数据有很多技术差异,即使在完成上述所有操作之后,这些差异也不会去除。
或者,您的主要兴趣可能是寻找细胞间表达的连续变化(例如,伪时间)。在这些情况下,别无选择,只能使用自动批次效应矫正工具。
但希望您可以尝试一下上面的方法去除,而不是默认运行批次效应矫正直接去除。