为了让教程具有较好的可重复性,我们直接使用 http://bioconductor.org/packages/release/bioc/html/AUCell.html 的案例即可。
只需要AUCell包的AUCell_calcAUC函数,就可以计算每个细胞的每个基因集的活性程度。难点在于如何制作输入数据,以及对结果的解读。
首先需要细胞的表达矩阵
AUCell包的教程使用了 GSE60361 数据集的单细胞转录组表达矩阵,是直接读取文本文件文件,代码具有学习价值,如下:
library(GEOquery)
attemptsLeft <- 20
while(attemptsLeft>0)
{
geoFile <- tryCatch(getGEOSuppFiles("GSE60361", makeDirectory=FALSE), error=identity)
if(methods::is(geoFile,"error"))
{
attemptsLeft <- attemptsLeft-1
Sys.sleep(5)
}
else
attemptsLeft <- 0
}
gzFile <- grep(".txt.gz", basename(rownames(geoFile)), fixed=TRUE, value=TRUE)
message(gzFile)
txtFile <- gsub(".gz", "", gzFile, fixed=TRUE)
message(txtFile)
gunzip(filename=gzFile, destname=txtFile, remove=TRUE)
library(data.table)
geoData <- fread(txtFile, sep="\t")
geneNames <- unname(unlist(geoData[,1, with=FALSE]))
exprMatrix <- as.matrix(geoData[,-1, with=FALSE])
rm(geoData)
dim(exprMatrix)
rownames(exprMatrix) <- geneNames
exprMatrix[1:5,1:4]
# Remove file
file.remove(txtFile)
# Save for future use
mouseBrainExprMatrix <- exprMatrix
save(mouseBrainExprMatrix, file="exprMatrix_AUCellVignette_MouseBrain.RData")
当然啦, 每个人写代码风格不一样,我就不会这样写。
最后读入的表达矩阵被整理好了,是小鼠的约2万个基因的3千多个细胞的表达矩阵,如下所示:
> dim(exprMatrix)
[1] 19972 3005
>
> exprMatrix[1:5,1:4]
1772071015_C02 1772071017_G12 1772071017_A05 1772071014_B06
Tspan12 0 0 0 3
Tshz1 3 1 0 2
Fnbp1l 3 1 6 4
Adamts15 0 0 0 0
Cldn12 1 1 1 0
一定要看清楚哦,是小鼠的约2万个基因的3千多个细胞的表达矩阵,这个原始的表达矩阵是reads的数量,但是整个分析并不关心具体的表达量是多少,只需要基因的排名即可,所以加上一个排序操作:
load("exprMatrix_AUCellVignette_MouseBrain.RData")
set.seed(333)
exprMatrix <- mouseBrainExprMatrix[sample(rownames(mouseBrainExprMatrix), 5000),]
dim(exprMatrix)
library(AUCell)
cells_rankings <- AUCell_buildRankings(exprMatrix, nCores=1, plotStats=TRUE)
cells_rankings
而且,考虑到计算量的问题,这里仅仅是随机抽样了5000个基因进行后续分析,如下:
> cells_rankings
Ranking for 5000 genes (rows) and 3005 cells (columns).
Top-left corner of the ranking:
cells
genes 1772071015_C02 1772071017_G12 1772071017_A05 1772071014_B06 1772067065_H06
0610009B22Rik 4408 1096 683 736 2780
0610009L18Rik 2406 1508 2212 4387 4402
0610010B08Rik_loc6 4468 4843 4048 4551 4552
0610011F06Rik 387 1111 1246 1533 817
0610012H03Rik 4867 2102 2245 4026 2405
0610039K10Rik 2051 1382 2213 3292 3753
Quantiles for the number of genes detected by cell:
min 1% 5% 10% 50% 100%
191 270 369 445 929 2062
后面的AUCell_calcAUC函数运行的时候,需要的就是制作好的cells_rankings变量。
然后需要基因集
通常是,我们是介绍msigdb数据库,MSigDB(Molecular Signatures Database)数据库中定义了已知的基因集合:http://software.broadinstitute.org/gsea/msigdb 包括H和C1-C7八个系列(Collection),每个系列分别是:
- H: hallmark gene sets (癌症)特征基因集合,共50组,最常用;
- C1: positional gene sets 位置基因集合,根据染色体位置,共326个,用的很少;
- C2: curated gene sets:(专家)校验基因集合,基于通路、文献等:
- C3: motif gene sets:模式基因集合,主要包括microRNA和转录因子靶基因两部分
- C4: computational gene sets:计算基因集合,通过挖掘癌症相关芯片数据定义的基因集合;
- C5: GO gene sets:Gene Ontology 基因本体论,包括BP(生物学过程biological process,细胞原件cellular component和分子功能molecular function三部分)
- C6: oncogenic signatures:癌症特征基因集合,大部分来源于NCBI GEO 发表芯片数据
- C7: immunologic signatures: 免疫相关基因集合。
不过,这个教程使用的是 Lavin, Y., et al. (2014) Tissue-Resident Macrophage Enhancer Landscapes Are Shaped by the Local Microenvironment. Cell 159, 1312–1326. doi: 10.1016/j.cell.2014.11.018 这个文章的数据的6个基因集 。
library(AUCell)
library(GSEABase)
gmtFile <- paste(file.path(system.file('examples', package='AUCell')), "geneSignatures.gmt", sep="/")
gmtFile
geneSets <- getGmt(gmtFile)
geneSets
names(geneSets)
geneSets <- subsetGeneSets(geneSets, rownames(exprMatrix))
cbind(nGenes(geneSets))
geneSets <- setGeneSetNames(geneSets,
newNames=paste(names(geneSets), " (", nGenes(geneSets) ,"g)", sep="")
)
geneSets
names(geneSets)
为了让这个基因集具有可比性,作者还加上了两个随机的基因集,以及一个管家基因的集合,代码如下:
## ----hkGs---------------------------------------------------------------------
# Random
set.seed(321)
extraGeneSets <- c(
GeneSet(sample(rownames(exprMatrix), 50), setName="Random (50g)"),
GeneSet(sample(rownames(exprMatrix), 500), setName="Random (500g)"))
countsPerGene <- apply(exprMatrix, 1, function(x) sum(x>0))
# Housekeeping-like
extraGeneSets <- c(extraGeneSets,
GeneSet(sample(names(countsPerGene)[which(countsPerGene>quantile(countsPerGene, probs=.95))], 100),
setName="HK-like (100g)"))
geneSets <- GeneSetCollection(c(geneSets,extraGeneSets))
names(geneSets)
最后是9个基因集,名字如下:
> names(geneSets)
[1] "Astrocyte_Cahoy (538g)" "Neuron_Cahoy (384g)" "Oligodendrocyte_Cahoy (477g)"
[4] "Astrocyte_Lein (7g)" "Neuron_Lein (13g)" "Microglia_lavin (165g)"
[7] "Random (50g)" "Random (500g)" "HK-like (100g)"
最后运行AUCell_calcAUC函数即可
代码如下:
cells_AUC <- AUCell_calcAUC(geneSets, cells_rankings)
cells_AUC
names(geneSets)
得到了结果:
> cells_AUC
AUC for 9 gene sets (rows) and 3005 cells (columns).
Top-left corner of the AUC matrix:
cells
gene sets 1772071015_C02 1772071017_G12 1772071017_A05 1772071014_B06 1772067065_H06
Astrocyte_Cahoy (538g) 0.1480482 0.1295743 0.15575904 0.16414458 0.14685944
Neuron_Cahoy (384g) 0.2726747 0.2889639 0.29359036 0.31331727 0.32973494
Oligodendrocyte_Cahoy (477g) 0.1549880 0.1291888 0.13828112 0.12526908 0.13452209
Astrocyte_Lein (7g) 0.1527294 0.1399535 0.21544715 0.24274100 0.10162602
Neuron_Lein (13g) 0.3725863 0.4251345 0.50997151 0.46818613 0.46533713
Microglia_lavin (165g) 0.0916712 0.0596262 0.06568681 0.08372346 0.07552168
就是我们给定的9个基因集,在全部的3000多个细胞的AUC值,可以简单理解为活性程度。
需要解释的2个点
首先是cells_rankings变量,每个细胞里面的基因都排序了,这个非常简单。需要注意的是 Top-left corner of the ranking,而且还计算了Quantiles for the number of genes detected by cell。
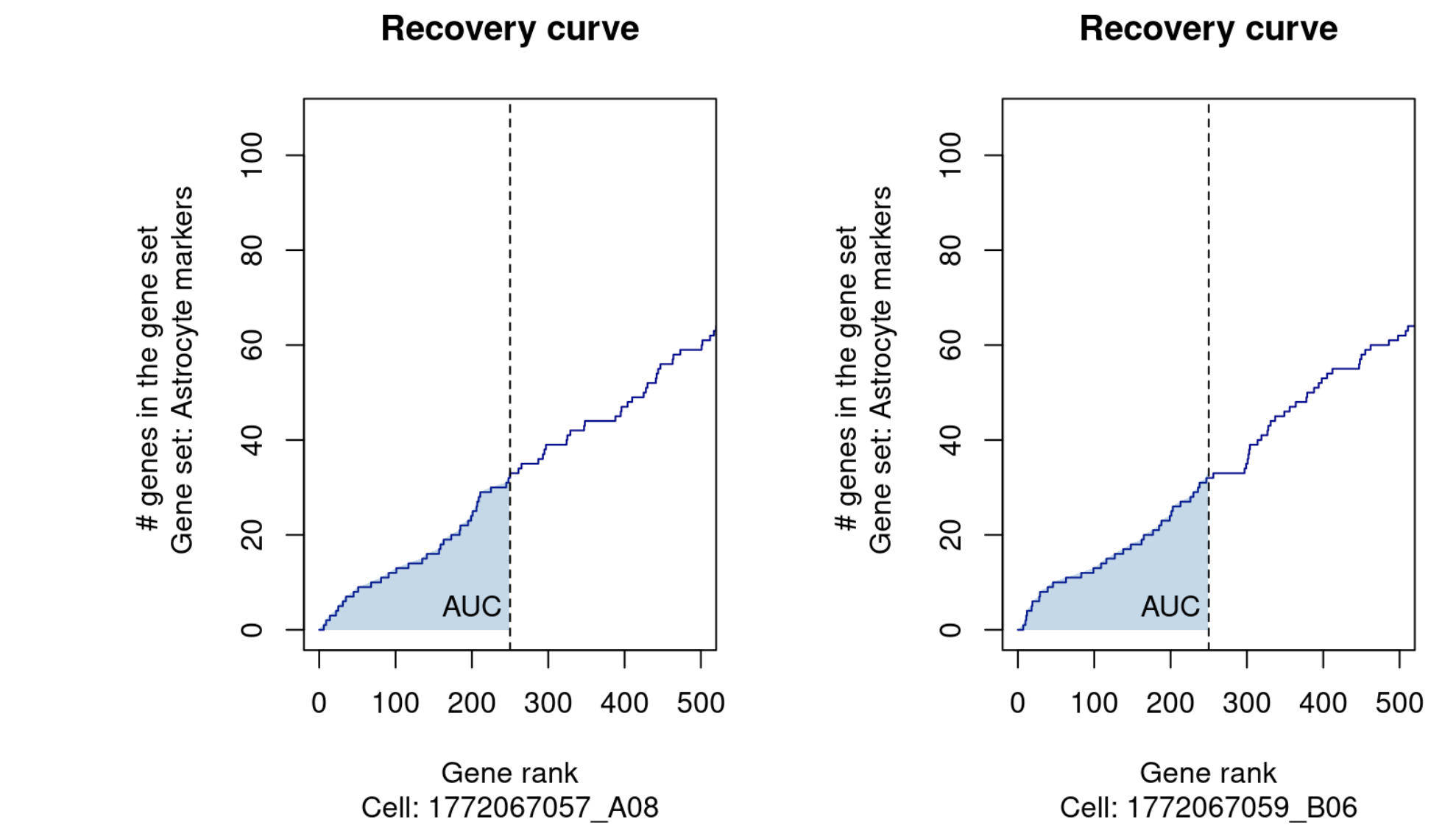
另外一个难点是Calculate the Area Under the Curve (AUC)的计算,作者给出来了第一个基因集在3000多个细胞里面随机挑选2个计算AUC的代码,如下:
geneSet <- geneSets[[1]]
geneSet
gSetRanks <- cells_rankings[geneIds(geneSet),]
par(mfrow=c(1,2))
set.seed(222)
aucMaxRank <- nrow(cells_rankings)*0.05
aucMaxRank
na <- sapply(sample(1:3005, 2), function(i){
x <- sort(getRanking(gSetRanks[,i]))
aucCurve <- cbind(c(0, x, nrow(cells_rankings)), c(0:length(x), length(x)))
op <- par(mar=c(5, 6, 4, 2) + 0.1)
plot(aucCurve,
type="s", col="darkblue", lwd=1,
xlab="Gene rank", ylab="# genes in the gene set \n Gene set: Astrocyte markers",
xlim=c(0, aucMaxRank*2), ylim=c(0, nGenes(geneSet)*.20),
main="Recovery curve",
sub=paste("Cell:", colnames(gSetRanks)[i]))
aucShade <- aucCurve[which(aucCurve[,1] < aucMaxRank),]
aucShade <- rbind(aucShade, c(aucMaxRank, nrow(aucShade)))
aucShade[,1] <- aucShade[,1]-1
aucShade <- rbind(aucShade, c(max(aucShade),0))
polygon(aucShade, col="#0066aa40", border=FALSE)
abline(v=aucMaxRank, lty=2)
text(aucMaxRank-50, 5, "AUC")
})
可以看到只需要考虑表达矩阵的top5%的基因即可,这个案例里面的是5000个基因的表达矩阵,所以只需要考虑250个基因。这250基因如果有位于我们感兴趣基因集里面纵坐标就加1,可以看到对第一个基因来说,可以加到约30个基因,这个基因集的数量是500,所以呢,其实top5%的基因里面哪怕是随机也是可以拿到25个基因的。
比如我们看第8个基因集,就是 “Random (500g)” ,纵坐标就到了25个基因。
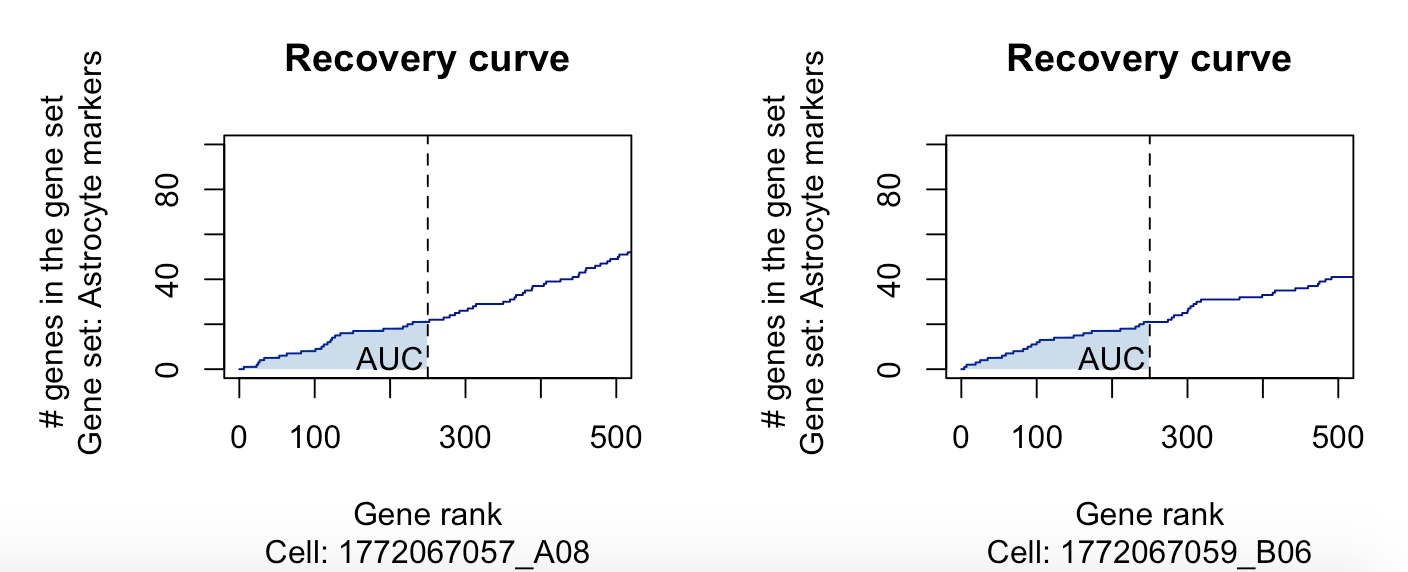
这个AUC就很容易理解啦,那个阴影部分的面积,至于如何计算呢,哈哈,这是一个数学问题。
结果图表的多样化
首先,我们可以拿到任意基因集的全部细胞的AUC值,这个呢,你可以理解为ssGSEA值,或者其它容易统计学指标。
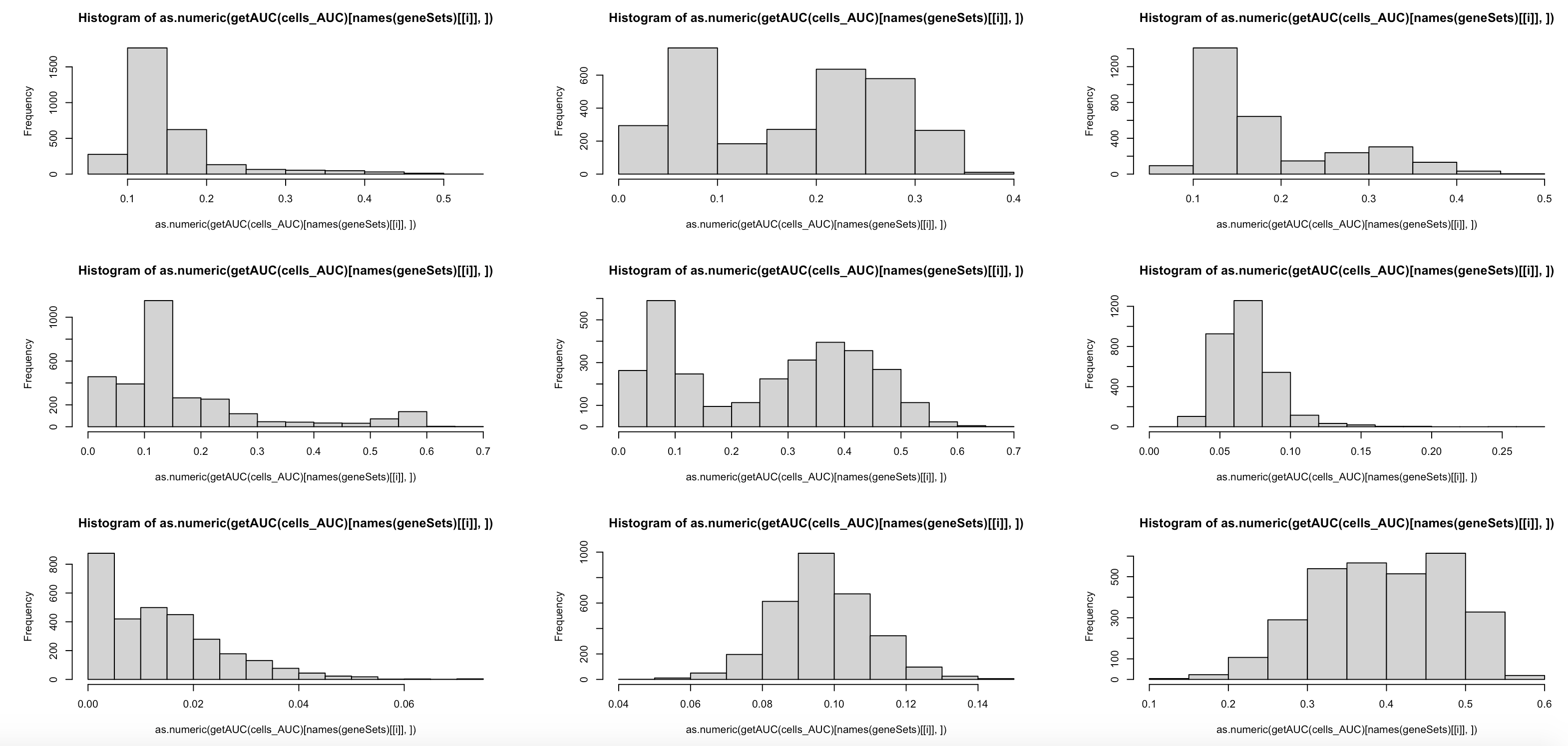
geneSet <- names(geneSets)[[8]]
aucs <- as.numeric(getAUC(cells_AUC)[geneSet, ])
hist(aucs)
set.seed(123)
par(mfrow=c(3,3))
# 这个代码稍微有点复杂,需要认真理解
lapply(1:9, function(i) print(hist(as.numeric(getAUC(cells_AUC)[ names(geneSets)[[i]], ]) )))
可以看到9个基因集在全部单细胞的AUC值分布范围:
然后作者提供了一个二值化的函数,根据AUC值的分布:
set.seed(123)
par(mfrow=c(3,3))
cells_assignment <- AUCell_exploreThresholds(cells_AUC, plotHist=TRUE, assign=TRUE)
warningMsg <- sapply(cells_assignment, function(x) x$aucThr$comment)
warningMsg[which(warningMsg!="")]
结果如下:
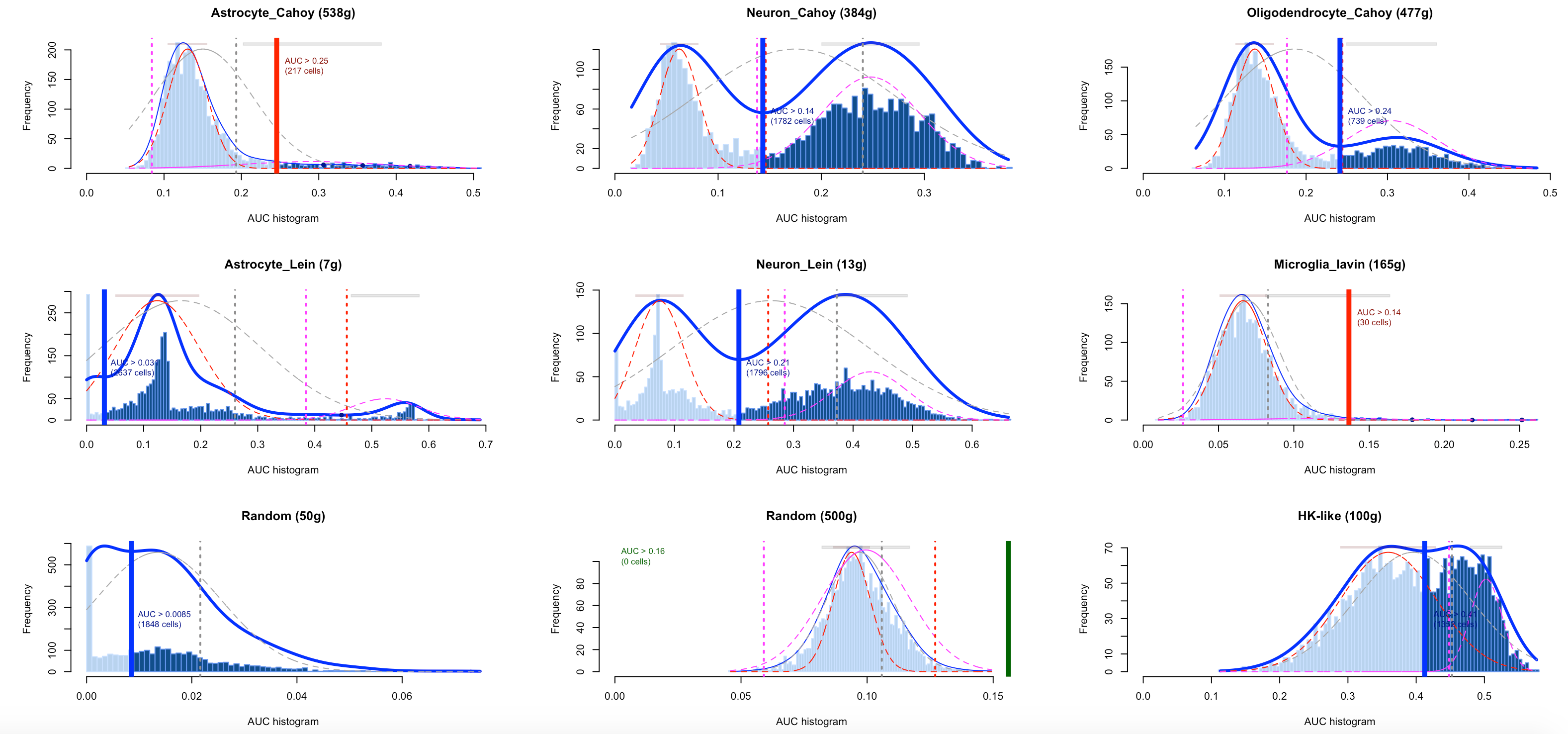
可以看到,每个基因集在全部的单细胞的AUC值都列出来了,但是呢,每个基因集的AUC阈值不一样。
根据各自基因集的阈值,把上面的结果变成二值化矩阵,代码如下:
names(cells_assignment)
cellsAssigned <- lapply(cells_assignment, function(x) x$assignment)
assignmentTable <- reshape2::melt(cellsAssigned, value.name="cell")
colnames(assignmentTable)[2] <- "geneSet"
head(assignmentTable)
dim(assignmentTable)
assignmentMat <- table(assignmentTable[,"geneSet"], assignmentTable[,"cell"])
dim(assignmentMat)
assignmentMat[,1:2]
apply(assignmentMat, 1, table)
结果如下:
> t(apply(assignmentMat, 1, table))
0 1
Astrocyte_Cahoy (538g) 2785 217
Astrocyte_Lein (7g) 365 2637
HK-like (100g) 1650 1352
Microglia_lavin (165g) 2972 30
Neuron_Cahoy (384g) 1220 1782
Neuron_Lein (13g) 1206 1796
Oligodendrocyte_Cahoy (477g) 2263 739
Random (50g) 1154 1848
>
这个二值化矩阵可以绘制热图,如下:
set.seed(123)
miniAssigMat <- assignmentMat[,sample(1:ncol(assignmentMat),100)]
library(NMF)
par(mfrow=c(1,1))
dev.new()
aheatmap(miniAssigMat, scale="none", color="black", legend=FALSE)
超级方便的可视化:

热图可视化还有更多方式,这里就不一一介绍啦。
把AUC值当做是一个表型信息
后续分析就可以是无穷无尽了,每个细胞都有线粒体基因含量百分比,有基因数量,有文库大小,这些表型信息是有限的,但是基因集是无限的, 每个基因集都可以计算一个AUC值,成为一个全新的表型信息。
以前的小提琴图,tSNE图展现线粒体基因含量百分比的,现在都可以用来展现这个AUC值。