两个月前的一个学徒作业:绘图本身很简单但是获取数据很难,完成率超级低,仅仅接到了不到十个邮件,而且有3个人做的是错的!!超级尴尬,其中有一个错误很明显,就是自以为是的排序,然后比对肿瘤组织和配对的正常组织的表达量,其实呢,排序错误会导致配对失败。
下面让优秀学徒具体讲解一下:
rm(list = ls())
options(stringsAsFactors = F)
library(ggstatsplot)
load('dat_input22.Rdata')
目前我的 dat 数据是这样的,可以看到同一个病人是有肿瘤组织和配对的正常组织的表达量的,而且呢,理论上是每一行一个样品的表达量信息:

对 pid 这一列排序后,group 这一列应该是相对应的奇数时是肿瘤组,偶数正常组。这时候就出现了问题,排列的没有规律性,如下:
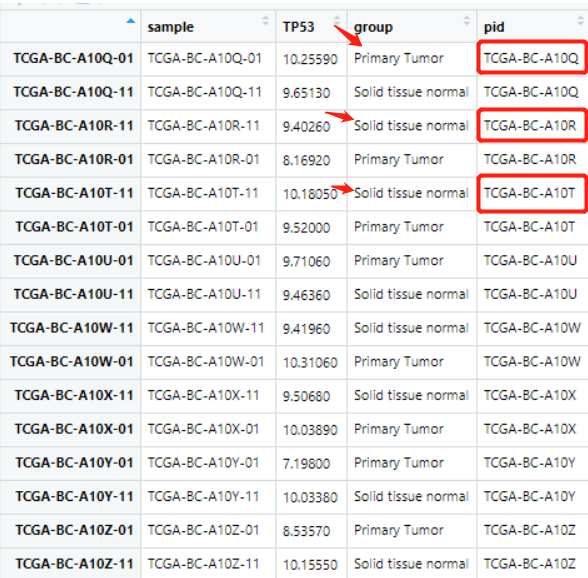
后面的数据就无法取,于是思考了一下两列的排序问题。(起初我并没有想到这一点,而是采用了其它复杂的方法完成了这个目标。但是jimmy老师点醒了我:凡是Excel能实现的数据操作,理论上R语言也可以,其实就是按照两列元素进行排序)
本来就只是一个简单的排序问题,随便搜搜就会有很好的答案,例如这样
df = dat
df = df[order(df[,4],df[,3],decreasing=TRUE),]
也就是说上面的代码呢,首先按照第4列排完序了,然后再来排一下第3列,我的数据也就得到了解决。
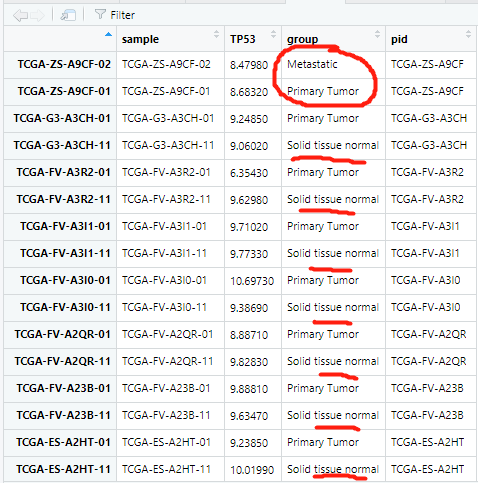
排列的整整齐齐:

并且后续的分析只需要在正常组和原位肿瘤组织中,不需要转移的肿瘤的这两个数据,应该删掉就行:
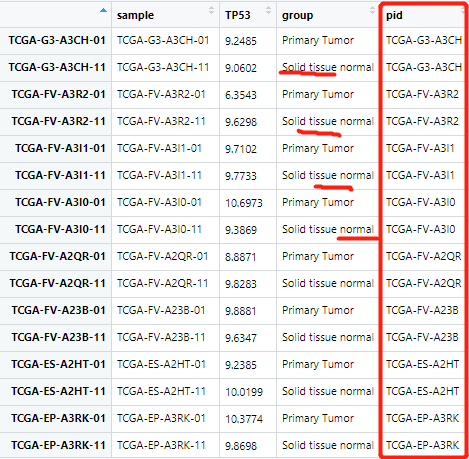
删除了多余的转移肿瘤的数据之后其实就完美了(都是那多出来的四个数据的问题,不然,第一次按照一列排序就可以很好)
之后就可以分别取出肿瘤样本和正常样本对应的 TP53 的表达量:
d=cbind(d[seq(1,nrow(d),by=2),],d[seq(2,nrow(d),by=2),])
identical(dat[,4],dat[,8])
head(dat)
normal=dat[,6];tumor=dat[,2]
这时候我的数据就结束了。
但是,
人们有时候真是有些奇奇怪怪的要求,索性研究一下排序问题。
哈哈,其实没有了,排序哪有什么问题,没有遇到具体问题时,哪会有排序的需求。下面还是让jimmy老师给大家讲解排序的需求吧:
扩展 筛选基因
我们读取一个表达矩阵文件,这个 GSE75688_GEO_processed_Breast_Cancer_raw_TPM_matrix.txt.gz 你应该是知道如何下载的。
rm(list=ls())
options(stringsAsFactors = F)
a=read.table('GSE75688_GEO_processed_Breast_Cancer_raw_TPM_matrix.txt.gz',
header = T)
a[1:4,1:4]
tail(a[,1:4])
表达矩阵如下:
> a[1:4,1:4]
gene_id gene_name gene_type BC01_Pooled
1 ENSG00000000003.10 TSPAN6 protein_coding 2.33
2 ENSG00000000005.5 TNMD protein_coding 0.00
3 ENSG00000000419.8 DPM1 protein_coding 60.70
4 ENSG00000000457.9 SCYL3 protein_coding 47.93
> tail(a[,1:4])
gene_id gene_name gene_type BC01_Pooled
57910 ERCC-00168 ERCC-00168 ERCC 0.00
57911 ERCC-00170 ERCC-00170 ERCC 0.00
57912 ERCC-00171 ERCC-00171 ERCC 0.00
57913 SPIKE1 EC2 SPIKE_IN 14940.70
57914 SPIKE2 EC15 SPIKE_IN 985.82
57915 SPIKE3 EC18 SPIKE_IN 0.00
可以看到前面的3列是基因信息,后面的才是表达矩阵的各个样品表达量信息。
ids=a[,1:3]
head(ids)
a=a[,4:ncol(a)]
a[1:4,1:4]
kp=rowSums(a>1) > 20
table(kp)
a=a[kp,]
ids=ids[kp,]
rownames(a)=ids[,1]
a[1:4,1:4]
虽然说第一列是ensembl的基因ID,第二列是我们想要的基因symbol。如果这个时候,我们强行把 rownames(a)=ids[,2] 就会报错,如下:

可以看到有大量的基因出现了多次,因为它们其实对应着不同的ensembl的基因ID,但是我们最后仍然是想要基因symbol。这个时候,我们就可以应用起来了我们的两列排序技巧:
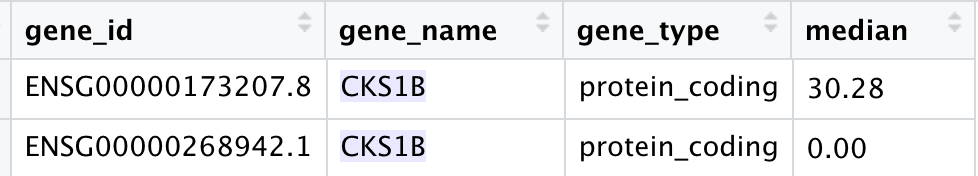
可以看到, 我们的ids数据框,首先是按照基因的symbol排序了,然后按照基因表达量排序了,所以可以简单的去冗余就拿到了合适的基因。全部的代码如下:
ids$median=apply(a,1,median)
ids=ids[order(ids$gene_name,ids$median,decreasing = T),]
ids=ids[!duplicated(ids$gene_name),]
a=a[ids$gene_id,]
rownames(a)=ids[,2]
这样,你的表达量矩阵,就是以唯一的基因symbol命名的啦。
虽然说两个不同的ensembl的基因ID,对应着同样的基因symbol,但是我们的挑选策略是,仅仅是保留表达量大的那个ensembl的基因ID。
如果你要问为什么两个不同的ensembl的基因ID会对应着同样的基因symbol
只能说是因为id本来就不统一,而且基因数量那么多,是超出人类认知范围的!这些知识点统称为生物信息学背景知识咯,甚至可以写一本书:
为什么要转换id?
有多少种ID?
什么id权威?
id是一一对应的吗?
ID是什么生信组织维护?
id有版本吗?
id一定正确吗?
什么情况下选择什么id?
不同数据库下载的id对应表一定一样吗?